Nucleosome Positioning on Yeast Plasmids Is Determined Only by the Internal Signal of DNA Sequence Occupied by the Nucleosome
G. I. Kiryanov1*, L. V. Isaeva1, L. N. Kinzurashvili2, and M. G. Zacharova1
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow 119992, Russia; fax: (095) 931-7211; E-mail: gikmsu@mail.ru2School of Biology, Tbilisi State University, Tbilisi, Georgian Republic
* To whom correspondence should be addressed.
Received December 11, 2002; Revision received January 15, 2003
The possible role of border factors in determining the nucleosome positioning on a DNA sequence was investigated. To this end a family of recombinant plasmids based on Gal10Cyc1 promoter and neomycin phosphotransferase gene NPTII were created. A DNA sequence adjoining the GalCyc promoter was varied in these plasmids. Three nearly equally represented nucleosome positions on the GalCyc promoter were found. In the basal plasmid an FRT sequence adjoins the GalCyc promoter at the right. It contains an internal signal of multiple positioning. Its replacement with different DNA sequences does not affect nucleosome positioning on the GalCyc promoter. The nucleosome positioning on the GalCyc promoter does not depend on nucleosome positioning (or its absence) on adjoining sequences. The same is true for nucleosome positioning on FRT sequence. It was found also that nucleosomes' positioning on the NPTII gene and their mutual disposition, namely the spacing between neighboring nucleosomes (linker length) are determined by the location of positioning signals only. Generally the nucleosome positioning in our experimental model is determined solely by internal DNA sequence occupied by nucleosome. On the other hand, the action of this internal positioning signal does not extend to neighboring DNA sequences.
KEY WORDS: yeast plasmids, nucleosome positioning, border signals, neighbor sequence shifting
The fixed histone octamer position on an individual DNA sequence and non-random distribution of octamer-interacting DNA sequences are described as the nucleosome positioning phenomenon [1]. Such positioning is usually most strongly pronounced on gene regulatory regions, whereas on gene coding sequences it is less pronounced or absent. One of the key aspects of nucleosome positioning is the nature of the positioning signal. A lot of data has been accumulated by now suggesting the nucleosome positioning signal to be determined by some local DNA structure peculiarities that enable or limit the possibility of DNA supercoiling around a histone octamer [2].
The affinities of short DNA sequences (both genomic and synthetic) to histone octamer were shown by nucleosome reconstruction experiments to be quite variable [3, 4]. A number of DNA sequence motifs have been defined that enhance or on the contrary limit the possibility of such interactions [5-8]. The real situation in vivo appears to be much more complicated and probably involves some additional positioning signals.
Thus nucleosome positioning on the same DNA sequence in vivo and in vitro sometimes is not the same [7, 8]. The nucleosome positioning on a gene may vary upon changes in its functional state [4, 7, 9, 10].
Several hypothetical additional nucleosome positioning signals have been proposed. Nucleosome positioning may be affected by so-called border factors, i.e., signals localized outside of the DNA sequence harboring the histone octamer. Some unusual structure of neighboring DNA sequence may play the role of such border signal. Another possible candidate is a DNA site occupied by a high-affinity protein. Still another possibility is that a sharply positioned nucleosome plays the role of border signal for neighboring nucleosomes.
The possible role of additional nucleosome positioning signals could not be clearly established in classic mononucleosome reconstruction experiments. Their significance can hardly be defined also from studies of nucleosome positioning on extended DNA sequences in vivo.
To evaluate the possible significance of border DNA sequences in nucleosome positioning, we changed the environment of a DNA sequence possessing an internal positioning signal. To this end a series of yeast recombinant plasmids was created containing a GalCyc promoter and variable neighboring DNA sequences.
MATERIALS AND METHODS
Bacterial and yeast cultures and yeast transformation and growth conditions. We used the E. coli strain M109 (Promega, USA) and Saccharomyces cerevisiae strain 2805 (MATa, pep4::His3, prbL-sigma, can-1, Gal2, his3, ura 3-52) kindly provided by C. K. Pu (Sang-Ki Rhii, GERI, Korea).
The yeast cells were transformed with plasmids pUG Fneo1Delta, pUG Fneo1Delta-2, pUG Fneo Delta(1Delta-2), and YepMMS by a LiCl method [11].
Transformants were selected on synthetic medium SD (0.67% yeast nitrogen base without amino acids (Difco, USA), 2% glucose, 60 µg/ml leucine, 30 µg/ml lysine, 0.5% ammonium sulfate, 1.5% agar (Difco)) or a complete medium YPD (1% yeast extract, 2% peptone, 2% glucose, 1.5% agar) containing aminoglycoside antibiotic geneticin (G418) at 300-600 µg/ml. The yeast clones selected were inoculated into 100-200 ml of liquid medium YPD or YPGal (complete medium with 2% galactose) and grown for 16-18 h to OD600 = 0.8-1.2.
The yeast recombinant plasmids pUG Fneo1Delta and pUG Fneo1Delta-2 were constructed by S. Saveliev et al. [12]. These plasmids are the derivatives of a yeast shuttle vector Yepsec I containing an E. coli neomycin phosphotransferase II gene (NPTII) flanked with a complete recombinant signal FRT from S. cerevisiae 2-µ plasmid on one side and a partially deleted one on the other. NPTII is in the sense expressed orientation relative to Gal10Cyc1 promoter in pUG Fneo1Delta, whereas it is in the anti-sense orientation in pUG Fneo1Delta-2. To produce the plasmid pUG FneoDelta(1Delta-2) a 1.1 kb SmaI-SacI (Ecl 136 II) fragment was cut out from pUG Fneo1Delta-2 and the resulting blunt ends were re-ligated. The fragment cut out contains partially deleted FRT signal and DNA segment downstream of SV40 gene terminator (from pSV2 neo by subcloning at BglII/BamHI sites).
To produce a yeast plasmid YepMMS containing an insert of mouse satellite DNA monomer a sample of total mouse liver DNA was digested with a restriction endonuclease MvaÉ. The products of hydrolysis were fractionated by 1.5% agarose gel electrophoresis. A 234 bp fragment of satellite DNA monomer was eluted from the gel. Its 3´-ends were blunted with Klenow fragment and ligated to a SmaI-digested plasmid vector pUC18. The orientation of satellite DNA monomer inserts was determined by digestion with restriction endonuclease Sau96É. The Sau96É site of satellite DNA was found to be in a proximal position relative to HindÉÉÉ site of pUC18 polylinker in all produced clones of recombinant plasmid (denoted pMMS). A 260 bp BamHÉ-SacÉ fragment of pMMS was further cloned between respective sites of YepsecÉ to produce recombinant yeast plasmid YepMMS.
Production of spheroplasts. Chromatin hydrolysis with micrococcal nuclease. The yeast cells (1*109) were collected by centrifugation, washed with sterile water and buffer solution (10 mM Tris-HCl, pH 7.4, 1 M sorbitol, 0.5 mM beta-mercaptoethanol), transferred to 2 ml centrifuge tubes and suspended in 1 ml of the same buffer. To produce the spheroplasts a yeast lytic enzyme Lyticase (800 U/mg; Sigma, USA) was added to cell suspension at 20 mg/ml and the mixture was incubated at 30°C for 5-10 min with gentle stirring. All the following stages were according to a scheme proposed earlier [13]. The resulting spheroplasts were suspended in 1 ml of the following buffer solution: 1 M sorbitol, 50 mM NaCl, 10 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 1 mM CaCl2, 1 mM beta-mercaptoethanol, 0.5 mM spermidine, 0.1% Nonidet NP-40.
The spheroplast suspension was transferred to microcentrifuge tubes in 200 µl aliquots, micrococcal nuclease was added at 150-350 U/ml and tubes were incubated at 37°C for 5 min. One of the tubes was incubated without micrococcal nuclease. The reaction was stopped by the addition of 0.5 M EDTA to 25 mM and 10% SDS to 0.5%.
Isolation of nucleosomal DNA. DNA samples isolated from yeast spheroplasts after extensive chromatin hydrolysis were fractionated by electrophoresis through 2% low-melting point agarose gels run in 0.04 M Tris-acetate/0.002 M EDTA buffer. Gene Ruler TM 100 bp DNA Ladder Plus fragments (MBI Fermentas) were used as molecular weight markers.
DNA fragments between 200 and 100 bp were eluted [14], concentrated by ethanol precipitation, and solubilized in an appropriate volume of water.
Oligonucleotides and their end-labeling. The following set of oligonucleotides was used:
1) 5´-gag-cag-atc-cgc-cag-gcg-3´ [(-257)-(-240)]
2) 5´-ttc-tat-aga-cac-gca-aac-aca-a-3´ [(-54)-(-33)]
3) 5´-acc-ggt-caa-ttc-gag-ctc-gg-3´ [(-16)-5]
4) 5´-cta-ccc-gtg-ata-ttg-ctg-aa-3´ [279]
5) 5´-aat-gag-aaa-tac-aca-ctt-ta 3´ [42-61]
6) 5´-tcc-acg-tcc-tac-agt-gga-ca 3´ [111-130].
The oligonucleotide primers were 5´-end-labeled by [gamma-32P]ATP (Obninsk, Russia) and T4-polynucleotide kinase [14]. The labeled primers were used in primer extension reactions with nucleosomal DNA as well as for plasmid dsDNA sequencing by a Taq DNA polymerase method.
The nucleosome borders were mapped by a primer extension method. The amplification reaction mixture (25 µl) contained 0.1-0.2 pmol (300-600 ng) of nucleosomal DNA, prepared as described above, 2.5 µl of 10× PCR-buffer (500 mM KCl, 100 mM Tris-HCl, pH 8.3, 15 mM MgCl2, 0.1% gelatin, 5% NP-40), 3-5 pmol of labeled primer, 2.5 µl of 10× dNTP mixture, and 5 U of Taq DNA polymerase. The amplification was carried out for 30 cycles of 95°C (1 min)-55°C (1 min)-72°C (2 min) preceded by initial 2 min denaturation at 95°C and followed by precipitation of DNA with 96% ethanol during the night at -20°C. The DNA precipitate was solubilized in 5 µl of stop-solution (95% formamide, 0.1% bromophenol blue, 0.1% xylene cyanole) and incubated for 2 min in a boiling water bath. DNA fragments were separated by 6% polyacrylamide gel electrophoresis in an LKB 2010 Macrophor Sequencing System (Pharmacia, Sweden).
A DNA sequencing kit (Silex M, Russia) was used for plasmid dsDNA sequencing. The gels were exposed to X-ray film during the night. Radioautographs were scanned with an LKB Ultroscan XL Laser Densitometer (Pharmacia).
RESULTS AND DISCUSSION
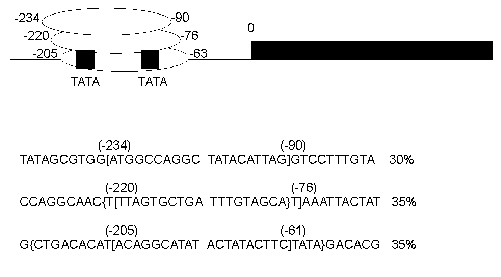
The schematic representation of analyzed (for nucleosome positions) DNA sequences is shown in Fig. 1. In the first plasmid construction (1Delta) a neomycin phosphotransferase gene (792 bp) (NPTII) is located downstream of a GalCyc promoter but is separated from this promoter by an insertion of FRT sequence (166 bp). FRT sequence is a signal recognized by FLP recombinase. We have mapped the nucleosome positions along this sequence both by an indirect end-labeling method [15] and by primer extension. In this paper we will describe nucleosome positioning on GalCyc promoter and nearby sequences. As shown in Fig. 2 the nucleosome is positioned on the GalCyc promoter sequence. There are three major nucleosome positions: from -234 to -90, from -220 to -76, from -205 to -61 (respective to 0-point--beginning of FRT sequence). These positions are nearly equally represented. Both TATA box sequences of the promoter are completely covered in all three positions.
Fig. 1. Scheme of analyzed parts of plasmids pUG Fneo1Delta, pUG Fneo1Delta-2, pUG FneoDelta(1Delta-2), and YepMMS. The complete FRT sequence (166 bp) is denoted by a thick long black half-arrow, the partially deleted FRT sequence (about 90 bp) by a short one, the NPTII gene (792 bp) by a hatched rectangle, and mouse satellite DNA monomer (234 bp) by a black rectangle; T is TATA boxes on the Gal10Cyc1 promoter (Cyc1, 250 bp). The restriction endonuclease cleavage sites are denoted by vertical arrows, and the oligonucleotides used for primer extension and replication direction by horizontal arrows.
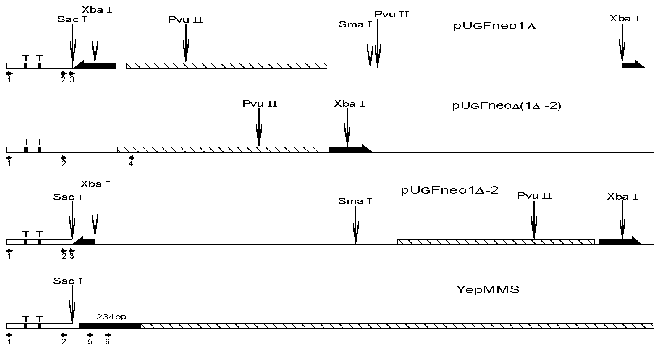
The first question to be solved was whether near-by sequences affect the nucleosome positioning on the GalCyc promoter. As can be seen in Fig. 1, the plasmid constructions used have different DNA sequences adjoining the GalCyc promoter. Namely, in the first plasmid (1Delta) it is an FRT sequence, in the plasmid Delta(1Delta-2) an inverted SV40 gene terminator sequence, in the plasmid (1Delta-2) a partially (by one half) deleted FRT sequence, and in the last plasmid presented a mouse satellite DNA monomer (234 bp). The three major nucleosome positions on GalCyc promoter are shown in Fig. 2. In none of the cases analyzed the nucleosome positioning on GalCyc promoter is affected by variations of adjoining DNA sequence. Moreover, it is not affected by the presence or absence of a positioned nucleosome on an adjoining DNA sequence. In particular, the presence of a multiply positioned nucleosome on the FRT sequence (see below) and regularly (every 10 bp) positioned nucleosome on mouse satellite DNA monomer do not affect nucleosome positioning on the GalCyc promoter.Fig. 2. Scheme of nucleosome positions on the DNA sequence of GalCyc promoter in all plasmid constructions. 0 is restriction endonuclease SacÉ cleavage site (Fig. 1); the brackets show the borders of nucleosome-occupied sequence. The figures show the nucleosome position relative to point 0; % is the relative part of each nucleosome position variant in the total nucleosome population.
On the other hand, the presence of positioned nucleosome on GalCyc promoter does not affect the nucleosome positioning on an adjoining FRT sequence either. The nucleosome positioning on the FRT sequence is not affected by change of its adjoining DNA sequence (see plasmid (1Delta-2) in Fig. 1) (data not shown).
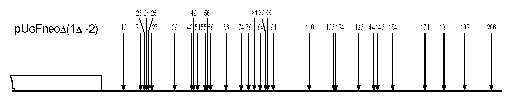
In plasmids 1Delta, (1Delta-2), and YepMMS the sequences adjoining GalCyc promoter possess some strong nucleosome positioning signals. One can suggest this to be a limiting factor in mutual nearby sequence interactions. The SV40 gene terminator sequence adjoining GalCyc promoter in the plasmid Delta(1Delta-2) on the contrary does not contain such signals, since there are quite a lot (more than 30) of nucleosome positions on these 200 bp DNA sequence (Fig. 3). Thus a sharply positioned nucleosome of GalCyc promoter does not serve as a positioning signal for the neighboring nucleosome.
The general conclusion is that, at least for the model used, border effects in nucleosome positioning are absent. Neither of the DNA sequences adjoining the GalCyc promoter affects its nucleosome positioning. Moreover, the presence of a sharply positioned or randomly localized nucleosome on an adjoining sequence do not serve as additional border signals in nucleosome positioning. Hence, in this model the nucleosome positioning is determined solely by an internal signal of nucleosome-occupied DNA sequence.Fig. 3. Scheme of DNA cleavage points in plasmid pUG Fneo Delta(1Delta-2). The arrows show the points of DNA cleavage upon digestion of chromatin with micrococcal nuclease, the figures their distance from point 0, and the square GalCyc promoter.
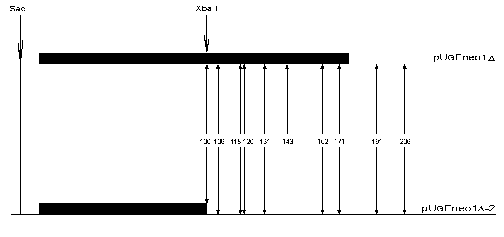
The nucleosome positioning on GalCyc promoter DNA sequence is sharp and lowly variable. Quite another example is nucleosome positioning on the FRT sequence (166 bp). We have found by a primer-extension method 17 differently represented variants of histone octamer positioning in this case. The nucleosome may shift along this sequence as far as by 90 bp towards the GalCyc promoter. In this position it is practically right up against the GalCyc promoter nucleosome which probably is the factor preventing further sliding. However, the factors limiting nucleosome sliding along FRT sequence in the opposite direction still remained unknown. To find the answer to this question we created a plasmid construction with a deletion of nearly half of the FRT sequence (Fig. 1). The neighborhood of this shortened FRT sequence is accordingly changed--it is adjoined downstream by an inversely oriented SV40 gene terminator sequence. A map of nucleosome positions on this sequence is shown in Fig. 4.
Ten DNA cleavage points corresponding to ten nucleosome border positions were found on the original sequence. These points are +100, +106, +118, +120, +131, +143, +162, +171, +191, and +206. Eight of them correspond to borders of nucleosomes located completely within FRT sequence, the other two of nucleosomes overlapping downstream neighbor sequence. Upon replacement of a part of FRT and adjoining sequence with quite a different one most of the cleavage points (9 of 10) remain (Fig. 4). In other words, the nucleosome positioning on FRT sequence overlapping the downstream adjoining sequence is totally determined by a positioning signal internal to the FRT sequence. In these cases the adjoining sequence (part of FRT + beginning of NPTII gene structural region or totally different inverted SV40 gene terminator sequence) is probably devoid of positioning signal. The limitation in nucleosome sliding (and hence the choice of positioning variant) in this case is also determined by internal positioning signals of histone octamer occupying DNA sequence itself.Fig. 4. Scheme of FRT nucleosome borders. The FRT sequence (166 bp) is denoted by the long black rectangle, the shortened FRT sequence (90 bp) by the short black rectangle. The DNA cleavage points corresponding to nucleosome borders are shown with vertical arrows. The figures show the border positions relative to restriction endonuclease SacI cleavage site.
The next case shows the absence of mutual influence of two neighboring nucleosomes positioned according to their internal positioning signals. The FRT sequence nucleosome was shown above to be capable of multiple positioning and sliding along DNA sequence for quite a long range (plasmid construction 1Delta). The neighboring (+2) NPTII gene nucleosome is sharply and rather invariably positioned. Its sliding along DNA sequences is limited to 30-40 bp. For all positioning variants these nucleosomes adjoining each other are rather widely spaced (about 100 bp). Such an unusually long internucleosome “linker” is determined solely by mutual positions of two positioning signals, i.e., by peculiarities of DNA sequences occupied by nucleosomes.
The general conclusion is that nucleosome positioning on DNA is determined solely by internal positioning signals of nucleosome-occupied sequence. The adjoining DNA sequences and adjoining positioned nucleosomes do not change the character of nucleosome positioning. The variability in nucleosome positioning found in some cases appears to be caused by factors affecting affinity of histone octamers to individual DNA sequence, such as competing proteins.
REFERENCES
1.Wolffe, A. P. (1994) Trends Biochem. Sci.,
19, 240-244.
2.Thoma, F., and Simpson, R. T. (1985) Nature,
315, 250-252.
3.Thaström, A., Lowary, P. T., Widlund, H. R.,
Cao, H., Kubista, M., and Widom, J. (1999) J. Mol. Biol.,
288, 213-229.
4.Widlund, H. R., Kuduvalli, P. N., Bengtsson, M.,
Cao, H., Tullins, T. D., and Kubista, M. J. (1999) J. Biol.
Chem., 274, 31847-31852.
5.Widlund, H. R., Cao, H., Simonsson, S., Magnusson,
E., Simonsson, T., Nielsen, P. E., Kahn, J. D., Crothers, D. M., and
Kubista, M. (1997) J. Mol. Biol., 267, 807-817.
6.Cao, H., Widlund, H. R., Simonsson, T., and
Kubista, M. (1998) J. Mol. Biol., 281, 253-260.
7.Linxweiler, W., and Horz, W. (1985) Cell,
42, 281-290.
8.Shen, C. N., and Clark, D. J. (2001) J. Mol.
Biol., 276, 35209-35216.
9.Moreira, J. M. A., and Holmberg, S. (1999) EMBO
J., 18, 2836-2844.
10.Cavalli, G., and Thoma, F. (1993) EMBO J.,
12, 4603-4613.
11.Rothstein, R. (1985) Cloning in Yeast.
DNA Cloning - a Practical Approach (Glover, G. M., ed.) Vol. II,
IRL Press, Oxford, pp. 45-66.
12.Saveliev, S. V., Fessing, M. Y., Kopylov, A. M.,
and Kirjanov, G. I. (1993) Curr. Genet., 24, 26-31.
13.Kent, N. A., and Mellor, J. (1995) Nucleic
Acids Res., 23, 3786-3787.
14.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning. A Laboratory Manual, Gold Spring
Harbor Laboratory Press, New York.
15.Nedospasov, S., and Georgiev, G. (1980)
Biochem. Biophys. Res. Commun., 92, 532-539.