Sialidase Activity in Normal and Atherosclerotic Human Aortic Intima
N. K. Golovanova*, E. V. Gracheva, O. P. Il'inskaya, E. M. Tararak, and N. V. Prokazova
Institute of Experimental Cardiology, Cardiology Research Center, Russian Ministry of Health, 3-ya Cherepkovskaya ul. 15a, Moscow, 121552 Russia; fax: (095) 415-2962; E-mail: golov@cardio.ru* To whom correspondence should be addressed.
Received June 3, 2002
Sialidase activity has been determined in homogenates of human aortic intima by measuring the amount of GM1 formed during the incubation of ganglioside GD1a with the tissue homogenates. Areas with atherosclerotic lesions as well as adjacent areas without histological evidence of atherosclerosis were taken for comparison. The rate of GM1 formation from GD1a in the presence of homogenates of the atherosclerotic intima was 20 pmol/h per mg protein. Homogenates of the unaffected intima did not desialylate GD1a. Sialidase activity of the atherosclerotic intima was linear for 1.5 h at GD1a content up to 1.5 nmol and at homogenate protein up to 1 µg. NH4Cl and NeuAc2en, inhibitors of lysosomal function and plasma membrane-bound sialidase, respectively, reduced sialidase activity of homogenates of the atherosclerotic intima by 94%. The results indicate that atherosclerotic lesions and unaffected intima differ in their activity and specificity of sialidases that cleave gangliosides.
KEY WORDS: sialidase activity, gangliosides, aortic intima, atherosclerosis
It is now well established that ganglioside levels in the intima of atherosclerotic plaque is several fold larger than that in unaffected intima. The glycosphingolipid composition is also altered in the process of atherogenesis [1-5]. The changes in glycosphingolipid composition and levels are considered to be a part of the pathobiological process in formation of atherosclerotic lesions [5, 6]. Nevertheless, metabolism of glycosphingolipids in smooth muscle vessels has not been studied yet.
Gangliosides are glycosphingolipids that contain one to several sialic acid residues and are integral component of the external side of plasma membranes of vertebrate cells. Gangliosides have been recognized as modulators of cellular functions such as cell growth and differentiation, cell adhesion, and cell-cell interactions [7-9]. The quantitative and qualitative composition of cell surface gangliosides plays an important role in cell behavior and results from the activity of sialyltransferases and sialidases, enzymes that attach and cleave, respectively, sialic acid. In the present work we performed a comparative study of sialidase activity in homogenates prepared from atherosclerotic and unaffected intimas of human aorta using ganglioside GD1a as an exogenous substrate.
MATERIALS AND METHODS
Gangliosides GM1 and GD1a were isolated from calf brain according to [10]. Chromatographic purity of gangliosides was assessed by TLC on high performance precoated silica gel plates (Merck, Germany) in the solvent system chloroform-methanol-0.25% calcium chloride (50 : 40 : 10) [10]. Cholera toxin B subunit conjugated to horseradish peroxidase (HRP-B), bovine serum albumin (BSA), 2-deoxy-2,3-dehydro-N-acetylneuraminic acid (NeuAc2en), buffer components, and detergents were purchased from Sigma (USA); 96-well microtiter plates were purchased from Biomedical (Russia).
Aortas from 25-65-year-old men and women were obtained aseptically within 12 h after death due to an accident, frozen in liquid nitrogen, and stored at -70°C until use. On the day of experiment the tissues were thawed, and further procedures were carried out at 4°C. The aortas were cleaned free of fat, cut lengthwise, and separated from adventitia. The occurrence and the type of atherosclerotic lesions were defined according to accepted classification [11]. Each individual aortal specimen was separated into unaffected (unlesioned) areas and areas with atherosclerotic lesions: atherosclerotic plaques or lipid streaks. The separated intima was blotted with filter paper, weighted, placed in 37.5 mM Tris-maleate buffer, pH 6.75, containing 0.25 M sucrose (3 ml buffer per 1 g tissue), homogenized in a Polytron homogenizer (Ultra-Turrax, Germany), and then centrifuged at 3,000g for 20 min. The resulting supernatant, a suspension of small particles (the homogenate after separation of rough pellet), was used for determination of sialidase activity. Protein concentration was determined by the method of Lowry-Peterson [12] using BSA for the calibration curve.
Sialidase activity toward GD1a was determined as described previously [13]. The incubation mixtures contained 0.22 mg/ml sodium cholate, 0.04 M acetate buffer, pH 4.2 (buffer A), 150 nmol GD1a, and 150 µg (as protein) intimal homogenate in a final volume of 450 µl. The incubations were carried out at 37°C. Buffer A, containing 250 µM NeuAc2en and 10 mM NH4Cl, was used in sialidase inhibition experiments. The incubations were stopped by addition of chloroform-methanol (2 : 1) mixture. The incubation mixtures were then evaporated and the dry residue was dissolved in 1 ml ethanol. In each experiment two control mixtures were identical to the incubation mixtures but did not contain the homogenate or GD1a. All experiments were conducted in duplicates.
The level of GM1 in homogenates of both intimal types before and after the incubation was determined in the same way except that GD1a was not added to the incubation mixture.
Quantitative measurements of GM1 formed were carried out by ELISA using HRP-B [14]. One hundred microliters of an ethanol extract of the incubation mixture was pipetted into wells of a microtiter plate and the plate was left at room temperature. Following complete evaporation of the solvent, 100 µl of 1% BSA was added to the wells. After incubation at 37°C for 1 h the solution was removed and the wells were washed with 0.001 M phosphate buffer, pH 7.2, containing 0.02% Tween-20 (buffer B). HRP-B was dissolved in buffer B (3 µl of subunit B per 20 ml) and 100 µl of this solution was pipetted into each well. The plate was incubated at 37°C for 1 h and the wells were washed with buffer B. Then 100 µl of o-phenylenediamine solution (8 mg/20 ml phosphate-citrate buffer) and 6 µl of 33% hydrogen peroxide solution were added. The color was allowed to develop for 30 min, and then 50 µl of 2.5 M H2SO4 was added. The amount of bound HRP-B was quantified colorimetrically with a Titertek Multiscan MCC spectrophotometer (USA) at 492 nm. The amount of GM1 formed was calculated from calibration curve that was developed using 0.004 to 2.0 pmol GM1 per well, with subsequent treatment with HRP-B as described above. The staining was linear with GM1 content up to 0.5 pmol per well. The accuracy of the method was ±5%.
To study the relationship between sialidase activity and intimal homogenate protein, GD1a concentration, or incubation time, the enzymatic reaction was carried out directly in microtiter wells. Due to high sensitivity of the method, a wide range of substrate (GD1a) concentrations were used depending on the activity of the intimal homogenate that was used in a particular experiment. When the dependence of sialidase activity on substrate concentration was studied, 0.25 to 2 nmol GD1a were introduced into each well. In kinetic experiments the lowest amount of GD1a per well was 1 nmol, whereas the relationship between activity and the amount of intimal homogenate protein was determined at 0.02 nmol GD1a per well. An ethanol solution of GD1a was left at room temperature until its complete evaporation. The intimal homogenate was introduced following by addition of other components of incubation mixture as described above. The incubations were performed at 37°C. Homogenate protein and incubation times are shown in legends for Figs. 1-3. The incubations were stopped by removing the liquid phase and subsequent washing with buffer B. Quantitative measurements of GM1 formed (adsorbed on plastic) were carried out by ELISA as described above.
RESULTS
Sialidase activity was determined in the homogenates prepared from the intima of five aortic specimens with atherosclerotic lesions. Adjacent areas with no signs of atherosclerosis were taken for comparison.
As can be seen from Table 1, incubation of homogenates of the atherosclerotic as well as normal intima with detergent resulted in accumulation of ganglioside GM1. The accumulated levels varied greatly from specimen to specimen (control column). When homogenates were incubated without GD1a, GM1 levels were always significantly lower in the atherosclerotic intima than in adjacent unaffected intima. The measurements of GM1 levels in unincubated intimal homogenates (Table 2) also showed lower levels of this ganglioside in the atherosclerotic intima.
Table 1. Sialidase activity (GM1, pmol/h per
mg protein) in the unaffected and atherosclerotic intimas of human
aorta
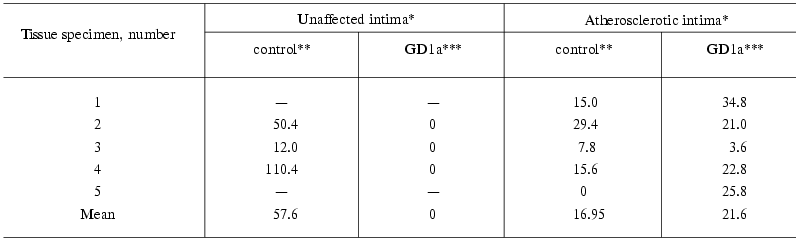
*Unaffected and atherosclerotic intimas were isolated from the same
aortic specimen, “-” means the absence of unlesioned areas in
the intima.
**Amount of GM1 formed in 1 mg homogenate during 1 h
incubation minus the amount of GM1 in the unincubated homogenate (see
Table 2).
***Amount of GM1 formed during 1 h incubation of the intimal homogenate
with GD1a as 1 nmol ganglioside per 1 µg homogenate
protein minus the amount of GM1 in the control experiment.
Table 2. GM1 levels (pmol/mg protein) in
homogenates of the unaffected and atherosclerotic intima
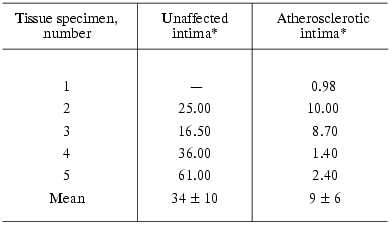
*Unaffected and atherosclerotic intimas were isolated from the same
aortic specimen, “-” means the absence of unlesioned areas in
the intima.
Incubation of homogenates of the atherosclerotic intima with GD1a resulted in formation of GM1, on the average, 20 pmol per 1 mg homogenate protein per 1 h (Table 1, GD1a column). The exception was one specimen whose activity was much lower than that of the other four. This specimen had also lower GM1 levels in control experiment (without addition of GD1a to the tissue homogenate) in comparison with the others. Homogenates of the unaffected intima did not desialylate GD1a (Table 1).
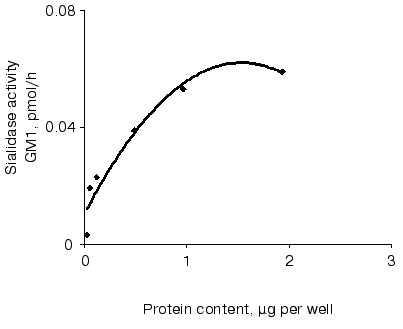
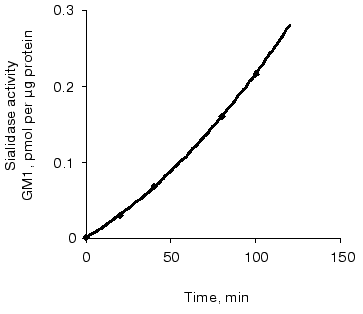
Sialidase activity of the atherosclerotic intima increased linearly with GD1a content up to 1.5 nmol (Fig. 1) and homogenate protein up to 1 µg (Fig. 2). The increase in GM1 was linear within 1.5 h when 1 nmol GD1a was treated with 1.3 µg homogenate protein of the atherosclerotic intima (Fig. 3).
Fig. 1. Relationship between sialidase activity of homogenates of the atherosclerotic intima and the amount of substrate GD1a. An ethanolic solution of GD1a (0.25 to 2 nmol per well) was placed into wells of a microtiter plate. After complete evaporation of the solvent, the homogenate (1.3 µg protein per well) was added and the plate was incubated for 1 h. The amount of GM1 formed was determined by ELISA. The wells without GD1a or the intimal homogenate served as controls.
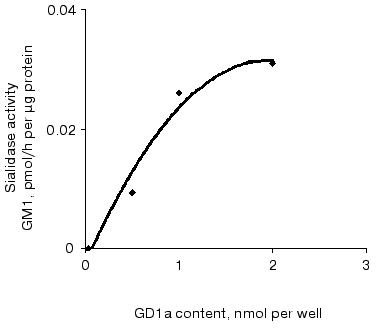
Fig. 2. Relationship between sialidase activity and homogenate protein of atherosclerotic intima. An ethanol solution of GD1a (0.02 nmol per well) was placed into wells. After complete evaporation of the solvent, the intimal homogenate (0.06 up to 2 µg protein per well) was added and the plate was incubated for 1 h. The amount of GM1 formed was determined by ELISA. Wells without GD1a or the intimal homogenate served as controls.
In the presence of NH4Cl and 2-deoxy-2,3-dehydro-N-acetylneuraminic acid, inhibitors of lysosomal function and plasma membrane sialidase, respectively, the formation of GM1 from GD1a in the presence of homogenates of the atherosclerotic intima decreased by 94%.Fig. 3. Relationship between sialidase activity of homogenates of the atherosclerotic intima and incubation time. An ethanol solution of GD1a (1 nmol per well) was placed into wells. After complete evaporation of the solvent, the intimal homogenate (1.3 µg protein per well) was added and the plate was incubated for 10-120 min. The amount of GM1 formed was determined by ELISA. Wells without GD1a or intimal homogenate served as controls.
DISCUSSION
Mammalian sialidases catalyze the first step of degradation of glycoconjugates by hydrolyzing ketoside bonds of sialic acid at the non-reducing terminal position of the carbohydrate chain. The levels of sialidases in tissues are very low and so the study of their enzymatic activity is determined by sensitivity of certain assay. In the present work a highly sensitive assay has been developed that is a modification of earlier reported method for analysis of sialidase activity toward gangliosides [14]. Desialylation of GD1a to GM1 catalyzed by sialidase was assessed by the developed assay in plastic microtiters for ELISA from the binding of HRP-B with monolayer of GM1 by subsequent colorimetric analysis of bound HRP-B with o-phenylenediamine and hydrogen peroxide. The sensitivity of this assay is 3*10-15 mol due to the great affinity of cholera toxin to ganglioside GM1.
The substrate specificity of sialidases from mammalian tissues is determined by the type of the bond of the terminal sialic acid residue (2-3, 2-8, 2-6) and does not depend on the structure of the glycoconjugate carbohydrate chain. For example, sialidases recently purified from plasma membranes of bovine and human brain were able to cleave the sialic acid from gangliosides GD1a, GD3, and GM3 with similar rates [15, 16]. Thus, despite the fact that GD1a is a minor component of gangliosides in the intima whereas GM3 and GD3 are its main constituents [4], the former can be used as a substrate to judge the total activity of tissue sialidase desialylating gangliosides.
In the course of the present studies it was found that the atherosclerotic intima in contrast to the unaffected intima had the ability to desialylate exogenous GD1a to GM1. The linear character of this desialylation on tissue homogenate protein, substrate concentration, and time as well as inhibition of desialylating activity by specific inhibitors [17], indicated that sialidases were responsible for the detected activity in homogenates of the atherosclerotic intima.
Several sialidases are found in mammalian cells and can be classified according to their subcellular localization into four types: cytosolic, lysosomal matrix, lysosomal membrane, and plasma membrane sialidase. Lysosomal and plasma membrane sialidases are best studied [15]. Cell gangliosides are mainly desialylated by membrane-bound sialidase, during which the particular orientation of glycoside molecules in the membrane bilayer plays a crucial role in cleavage efficiency of the membrane enzyme [18-20]. Glycoproteins are the preferential substrate for lysosomal sialidase, though gangliosides can also be desialylated by lysosomal sialidases in the presence of detergents [15, 21]. The levels of lysosomal sialidase in the unaffected intima seem not to be sufficient to detect its activity toward exogenous ganglioside substrate. By contrast, the activity of most lysosomal enzymes in atherosclerotic lesions are much higher than in the unaffected intima [22-25], mostly due to the influx of blood monocytes with their subsequent differentiation into macrophages and the dedifferentiation of smooth muscle cells from contractile to macrophagal phenotype as well [26-28]. Previously it was found that highly activated monocytes/macrophages were able to secrete lysosomal sialidases [29].
Thus, it can be suggested that the observed desialylation of GD1a by homogenates of the atherosclerotic intima in the presence of detergents is carried out by lysosomal sialidase. This is supported by the fact that sialidase activity of the atherosclerotic intima is lower than that of plasma membrane sialidase previously studied in fibroblasts and human neuroblastoma cells [14, 16-18]. In our experiments at substrate excess the desialylating activity was, on average, 3-fold lower than the accumulation of GM1 during the incubation of homogenates of the unaffected intima without exogenously added GD1a. This accumulation was more likely to be a result of desialylation by membrane sialidase of endogenous gangliosides GD1a, GD1b, and GT1b that were present in aortic tissue [2-4]. The estimation of kinetic parameters for this desialylation is problematic. Therefore, we cannot define the accumulation of GM1 during incubation of homogenates in the absence of GD1a as sialidase activity. Nevertheless, it should be noted that accumulation of GM1 occurred more intensively in the case of the unaffected intima.
This work was supported by the Russian Foundation for Basic Research (grant No. 02-04-48620) and Swiss National Science Foundation (Project 7SUP J062375). We thank Dr. N. N. Samovilova for help in preparation of the manuscript.
REFERENCES
1.Bobryshev, Y. V., Lord, R. S. A., Golovanova, N.
K., Gracheva, E. V., Zvezdina, N. D., and Prokazova, N. V. (2001)
Biochim. Biophys. Acta, 1535, 87-99.
2.Wen, F.-Q., Jabbar, A. A., Patel, D. A., Kazarian,
T., and Valentino, L. A. (1999) Arterioscler. Thromb. Vasc.
Biol., 19, 519-524.
3.Mukhin, D. N., Chao, F. F., and Kruth, H. (1995)
Arterioscler. Thromb. Vasc. Biol., 15, 1607-1615.
4.Prokazova, N. V., Orekhov, A. N., Mukhin, D. N.,
Mikhailenko, I. A., Kogtev, L. S., Sadovskaya, V. L., Golovanova, N.
K., and Bergelson, L. D. (1987) Eur. J. Biochem., 167,
349-352.
5.Prokazova, N. V., Golovanova, N. K., Gracheva, E.
V., Zvezdina, N. D., Bobryshev, Y. V., and Lord, R. S. A. (1999)
Angiol. Vasc. Surgery, 5, 151-165.
6.Chatterjee, S. (1998) Arterioscler. Thromb.
Vasc. Biol., 18, 1523-1533.
7.Tettamanti, G., and Riboni, L. (1993) Adv. Lipid
Res., 25, 235-267.
8.Hakomori, S., and Igarashi, Y. (1995) J.
Biochem., 118, 1091-1103.
9.Lloyd, K. O., and Furukawa, K. (1998)
Glycoconjugate J., 15, 627-636.
10.Bergelson, L. D., Dyatlovitskaya, E. V.,
Molotkovsky, Yu. G., Batrakov, S. G., Barsukov, L. I., and Prokazova,
N. V. (1981) Preparative Lipid Biochemistry [in Russian], Nauka,
Moscow, p. 209.
11.Smith, E. B., and Slater, R. S. (1972)
Atherosclerosis, 8, 229-242.
12.Peterson, G. L. (1977) Analyt. Biochem.,
83, 346-356.
13.Komleva, A. V., Prokazova, N. V., Gabrielyan, N.
D., Mukhin, D. N., Kashnikova, L. N., Zvezdina, N. D., Martinova, L.
E., and Panasyuk, A. F. (1995) Clin. Exp.
Rheumatol.,13,581-588.
14.Ogura, K., Ogura, M., Anderson, R. L., and
Sweely, C. C. (1992) Analyt. Biochem., 200, 52-57.
15.Bieberich, E., Liour, S. S., and Yu, R. K. (2000)
Meth. Enzymol., 312B, 339-358.
16.Kopitz, J., Sinz, K., Brossmer, R., and Cantz, M.
(1997) Eur. J. Biochem., 248, 527-534.
17.Kopitz, J., Mühl, C., Ehemann, V., Lehmann,
C., and Cantz, M. (1997) Eur. J. Cell Biol., 73, 1-9.
18.Kopitz, J., Oehler, C., and Cantz, M. (2001)
FEBS Lett., 491, 233-236.
19.Kalka, D., von Reitzenstein, C., Kopitz, J., and
Cantz, M. (2001) Biochem. Biophys. Res. Commun., 283,
989-993.
20.Scheider-Jakob, H. R., and Cantz, M. (1991)
Biol. Chem. Hoppe-Seyler,372, 443-450.
21.Beck, M., Scheuring, E., Voelter, H.-U., Brandt,
J., and Harzer, K. (1996) Clin. Chim. Acta, 251,
163-171.
22.Tararak, E. M., Zeltzer, G. L., and
Shaposhnikova, E. S. (1983) Kardiologiya, 23,72-77.
23.Berberian, P. A., and Jenison, M. W. (1985)
Exp. Mol. Pathol., 43, 22-35.
24.Ecsedi, G. G., Virag, S., and Hidvegi, E. J.
(1981) Atherosclerosis, 39, 183-90.
25.Tabargi, S. I., Feofilaktova, S. N., Varsanovich,
E. A., Vasil'ev, A. V., and Tutel'yan, V. A. (1990) Vopr. Med.
Khim.,36, 32-34.
26.Ross, R. (1999) New England J. Med.,
340, 115-126.
27.Flower, S., Shio, H., and Halley, N. J. (1979)
Lab. Invest., 41, 372-376.
28.Williams, K. J., and Tabas, I. (1995)
Arterioscler. Thromb. Vasc. Biol., 15, 551-561.
29.Pillatte, Y., Bigon, J., and Lamoré, C. R.
(1993) Glycobiology, 3, 201-217.