REVIEW: Inhibition of Enzymes of Polyamine Biosynthesis by Substrate-Like O-Substituted Hydroxylamines
A. R. Khomutov
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 119991 Russia; fax: (095) 135-1405; E-mail: alexkhom@genome.eimb.relarn.ru
Received May 23, 2002
The biogenic amines spermine, spermidine, and putrescine are essential factors of cell growth and differentiation. To inhibit pyridoxal-5´-phosphate dependent ornithine decarboxylase and pyruvate dependent S-adenosylmethionine decarboxylase, key enzymes of polyamine biosynthesis, a system of substrate-like O-substituted hydroxylamines is suggested. The best of these compounds were active at nanomolar concentrations. High potency and specificity of this type of inhibitors are discussed in terms of structural similarity of E-I and E-S complexes.
KEY WORDS: polyamines, spermine, spermidine, O-substituted hydroxylamines, S-adenosylmethionine decarboxylase, ornithine decarboxylase
The polyamines spermine (Spm) and spermidine (Spd) as well as their precursor putrescine (Put) are found in significant amounts in eukaryotic and prokaryotic cells, in plants, and in viruses. Polyamines are essential factors of cell growth and differentiation, while tumor cells have elevated levels of polyamines as compared to normal cells.
Polyamines exist at physiological pH as polycations, and they efficiently interact with DNA, RNA, and nucleoproteins, including ribosomes [1]. Polyamines are native regulators of the activities of topoisomerases and restrictases as well as enzymes of DNA and RNA biosynthesis. Spermidine, being the one donor of the aminobutyl fragment in the deoxyhypusine synthase reaction, is essential for post-translational modification of the epsilon-amino group of a Lys residue, located in a highly conservative region of the polypeptide chain, of initiation factor eIF-4A, which is present in all eukaryotes. Transformation of Lys into hypusine [Nepsilon-(4-amino-2-hydroxybutyl)-L-lysine] is crucial for the activity of eIF-4A [2]. Polyamines are involved in the regulation of mitochondrial Ca2+ transport [3], serve as modulators of NMDA receptor [4] and as effectors of K+ transport [5]. Recently, it was demonstrated that polyamines participate in apoptosis [6-8].
The totality of different known cellular functions of polyamines makes it possible to consider Spm and Spd as universal low molecular weight regulators of cellular metabolism. The decrease in intracellular polyamine content results in inhibition of cell growth or even cell death.
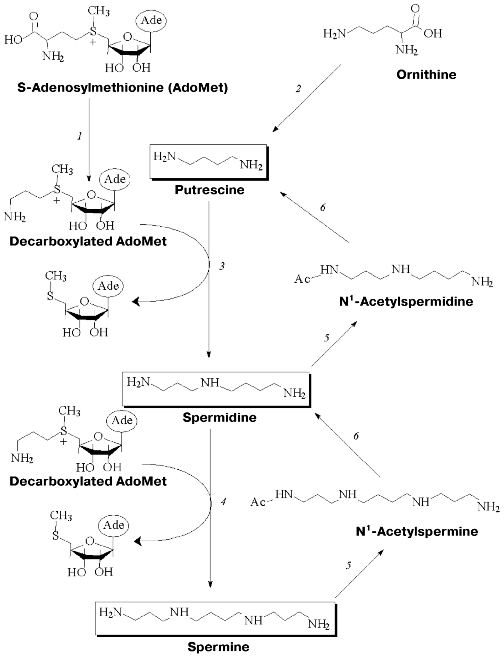
Polyamine metabolism is tightly related to transformations of ornithine, methionine, and S-adenosylmethionine (AdoMet). Key enzymes of polyamine biosynthesis are pyridoxal-5´-phosphate (PLP) dependent ornithine decarboxylase (ODC) and pyruvate-dependent S-adenosylmethionine decarboxylase (AdoMetDC), while spermine/spermidine-N1-acetyltransferase (SSAT) is the key enzyme of polyamine catabolism (Fig. 1). These key enzymes are short-living proteins (half-life in cells is about 30 min). Both biosynthesis and degradation of all three of them are easily induced. Polyamine metabolism is self-regulated. Cells are equipped with a system of active polyamine transport and this system makes an important contribution to intracellular polyamine content and regulation of their metabolism.
Specific regulation of polyamine metabolism is a rather complicated task. However, this approach is widely used to investigate cellular functions of polyamines. Specific inhibitors of polyamine biosynthesis and the compounds able to activate the enzymes of polyamine catabolism, as well as inhibitors of polyamine transport, have a wide spectrum of biological activities [9-12] that give practical importance to the investigation of polyamine metabolism and cellular functions.Fig. 1. Metabolism of polyamines: 1) S-adenosylmethionine decarboxylase(AdoMetDC); 2) ornithine decarboxylase (ODC); 3) spermidine synthase; 4) spermine synthase; 5) spermine/spermidine-N1-acetyltransferase (SSAT); 6) polyamine oxidase.
The step of ornithine and AdoMet decarboxylation involves the formation of a Schiff base between PLP or pyruvate and a substrate amino acid. Respectively, it was quite obvious to use carbonyl reagents (derivatives of hydrazine, hydroxylamine, semicarbazide, etc.) to inhibit these enzymes. Objectively, alpha-hydrazinoornithine effectively (IC50 0.5-2.0 µM) inhibited ODC, but the ease of oxidation of hydrazino-group decreased the value of this compound for cell experiments [13, 14]. Phenyhydrazine was found to be an irreversible inhibitor of AdoMetDC (IC50 0.5 mM) and the corresponding phenylhydrazone was identified [15]. Canaline (gamma-aminooxy-alpha-amino butyric acid) and aminooxyacetic acid turned to be not very active inhibitors (IC50 1-0.1 mM) of ODC [16] and AdoMetDC [17], respectively. So, application of simple carbonyl reagents could not obey the required specificity and effectiveness of the inhibition of ODC and AdoMetDC.
However, in the case of PLP-dependent decarboxylases of His [18] and Glu [19], as well as for GABA transaminase [20], the efficiency of inhibition of their enzymes by different O-substituted hydroxylamines depended on the structure of the alkyl substituent. Structural similarity between amino acid side-chain and alkyl fragment of O-substituted hydroxylamine was postulated to be important for effective inhibition of these PLP-dependent enzymes.
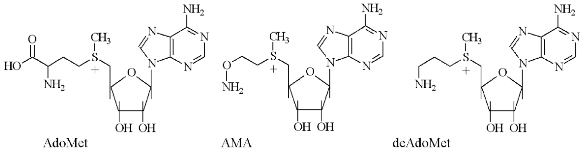
Respectively, for specific and irreversible inhibition of AdoMetDC a family of earlier unknown sulfonium-containing O-substituted hydroxylamines, which may be considered as analogs of decarboxylated AdoMet (deAdoMet) and methylmethionine, were designed and synthesized [21]. A key compound of this family was S-(5´-deoxy-5´-adenosyl)[(methylthio)ethyl]hydroxylamine--AMA (Fig. 2) [22].
AMA is an analog of deAdoMet, in which a methylene group, connected with the amino group, is substituted onto the oxygen atom, i.e., the aminomethylene group is converted into an aminooxy group, which was the driving force of irreversible inhibition. Such a substitution is isosteric and the similarity of the inhibitor with the substrate as well as specificity of AMA binding with the enzyme active center are not changed greatly, which is crucial for specificity of inhibition. The aminooxy group has pKa ~ 5.0, which is about five orders of magnitude lower than the pKa of the amino group of deAdoMet. Thus, at physiological pH AMA is monocation, which may be essential for the transportation of the analog into the cell.Fig. 2. Structures of S-adenosylmethionine (AdoMet), decarboxylated S-adenosylmethionine (deAdoMet), and S-(5´-deoxy-5´-adenosyl)[(methylthio)ethyl]hydroxylamine (AMA).
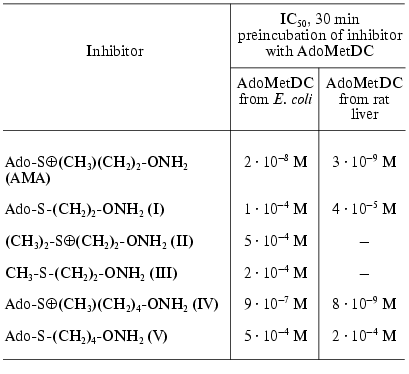
As depicted in Table 1, the effectiveness of inhibition was determined by the chemical structure of the inhibitor and the most effective compound (AMA) was isosteric with the product of enzymatic reaction--deAdoMet. The lack of the sulfonium center and adenosine group, these probably being the main fragments responsible for the recognition, immediately decreased the activity of the inhibitor by about five orders of magnitude (compare compound (I) and AMA, compound (II) and AMA). Nonprotonated H2NO-group formed the stable oxime with the pyruvate residue in the active center of AdoMetDC. Inhibition was irreversible and developed in time. Substrate was incapable of restoring the activity of the enzyme, but it competed with AMA for binding with the enzyme active center.
Table 1. Inhibition of AdoMetDC by
hydroxylamine-containing analogs of decarboxylated AdoMet [23]
AMA inhibited PLP-dependent ODC and aspartate aminotransferase only in µM and mM concentrations, respectively, that confirmed the specificity of the inhibitor towards AdoMetDC.
When synthesized, AMA turned out to be the first specific and highly effective irreversible inhibitor of AdoMetDC, acting in nM concentrations, which is 1000 times better than the best of the inhibitors known before.
The activity of most eukaryotic AdoMetDC is allosterically regulated by putrescine. The mechanism of this activation apparently includes putrescine-induced conformational changed of the protein structure, that results in 10-fold decrease in Km of the substrate (0.06 mM at pH 7.5), in changes in the pH profile of enzymatic reaction, and also in decrease in kcat to 2.5 sec-1 at 37°C [24]. On the contrary, bacteria, including E. coli, have Mg2+-dependent AdoMetDC. It was documented, that Mg2+ is essential for effective inhibition of AdoMet by AMA. In the absence of Mg2+, AMA was inactive even at 5.9 µM concentration (IC50 for the enzyme from E. coli was 0.002 µM, see above). Similar decrease in the activity in the absence of Mg2+ was observed also in the case of 3-aminooxy-1-aminopropane (APA), which was a nonspecific irreversible inhibitor of AdoMetDC. This may mean that Mg2+ causes conformational changes in the enzyme structure and catalytically important pyruvate residue become available and reactive towards amino group of the substrate as well as towards the aminooxy residue of AMA [25].
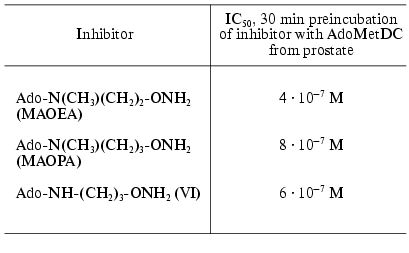
The second type of hydroxylamine containing inhibitors of AdoMetDC (Fig. 3) include 5´-deoxy-5´-[N-(gamma-aminooxypropylamino)]adenosine (VI) [26], 5´-deoxy-5´-[N-methyl-N-(beta-aminooxyethylamino)]adenosine (MAOEA), and 5´-deoxy-5´-[N-methyl-N-(gamma-aminooxypropylamino)]adenosine (MAOPA) [27].
The inhibitory activity of MAOEA towards AdoMetDC from prostate was for about 100 times weaker than AMA did in the case of rat liver enzyme (compare Tables 2 and 1). The activities of (VI), MAOPA, and MAOEA were within the same order of magnitude. In all cases inhibition was irreversible and developed in time. Substrate was incapable of restoring the activity of AdoMetDC, but it competed with all inhibitors for binding with enzyme active center.Fig. 3. Structures of 5´-deoxy-5´-[N-methyl-N-(gamma-amino-gamma-carboxypropyl)]-adenosine (aza-AdoMet); 5´-deoxy-5´-[N-methyl-N-(gamma-aminopropyl)]-adenosine (de-aza-AdoMet), and 5´-deoxy-5´-[N-methyl-N-(beta-aminooxyethylamino)]-adenosine (MAOEA).
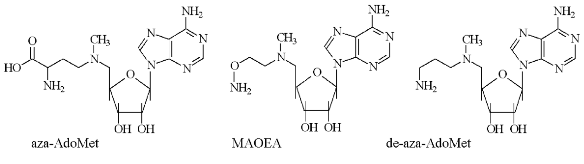
Table 2. Inhibition of AdoMetDC by
hydroxylamine-containing analogs of decarboxylated aza-AdoMet [26, 27]
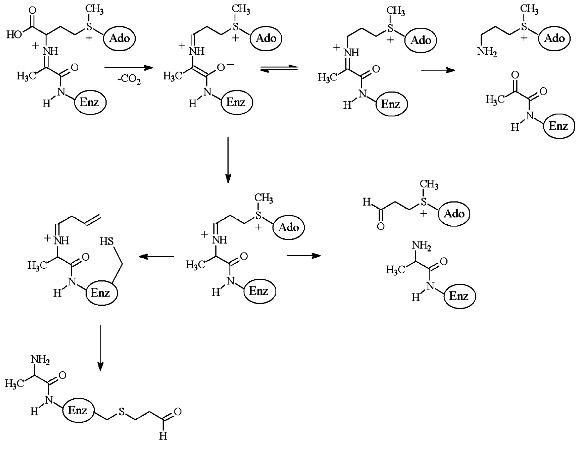
The peculiarities of the interaction of de-AdoMet and de-aza-AdoMet with AdoMetDC may explain observed differences in activities of AMA and MAOAE. Both de-AdoMet and de-aza-AdoMet were competitive inhibitors of AdoMetDC. However, only de-AdoMet formed a Schiff base with AdoMetDC, which can be reduced by NaBH3CN [28]. An interesting property of AdoMetDC is time-dependent inactivation of the enzyme as the result of the incubation with substrate, product, or alpha-methyl-AdoMet [28]. This was interpreted in terms of transamination of the enzyme and subsequent addition of Cys140 (enzyme from E. coli) to acrolein system [29-31] (Fig. 4). However, neither aza-AdoMet nor de-aza-AdoMet were capable of inhibiting the enzyme irreversibly [28]. Finally, nitrogen atom of aza-AdoMet has abnormally low pKa = 7.09 and the compound was able to substitute effectively for AdoMet, which is a corepressor of methionine repressor (MetJ) E. coli, only at pH 5.5, while at pH 7.4 the analog was not recognized as AdoMet [32]. So, it might be possible to assume that methylamino groups of MAOEA and MAOPA will also have low pKa and these compounds may be considered as analogs of decarboxylated S-adenosylhomocysteine, but not of decarboxylated AdoMet. Probably, only X-ray investigation of the corresponding E-I complexes will give an exact picture of the interaction of AMA and MAOEA with the active cite of AdoMetDC.
The second key enzyme of polyamine biosynthesis is PLP-dependent ODC, which is the most investigated enzyme of polyamine metabolism. Many different inhibitors of ODC are known, including suicide alpha,alpha-difluoromethyl ornithine (DFMO), which was synthesized in 1978 [33] and at present it is still one of the main and definitely most detailed investigated inhibitor of ODC both in vitro and in vivo [34].Fig. 4. Scheme of enzymatic decarboxylation of AdoMet and irreversible inhibition of AdoMetDC by the substrate (taken from [31]).
However, like the case of AdoMetDC, among substrate, or more exactly, among putrescine-like O-substituted hydroxylamines it was possible to find effective and specific inhibitors of ODC (Fig. 5). The first, simplest, and one of most active compound of this series was 3-aminooxy-1-aminopropane (APA) [35]. Inhibition activity of APA, as well as the activities of a number of it analogs, are given in Table 3.
Table 3. Inhibition of ODC by hydroxylamine-containing analogs of putrescine [35, 36]Fig. 5. Structures of putrescine and 3-aminooxy-1-aminopropane (APA).

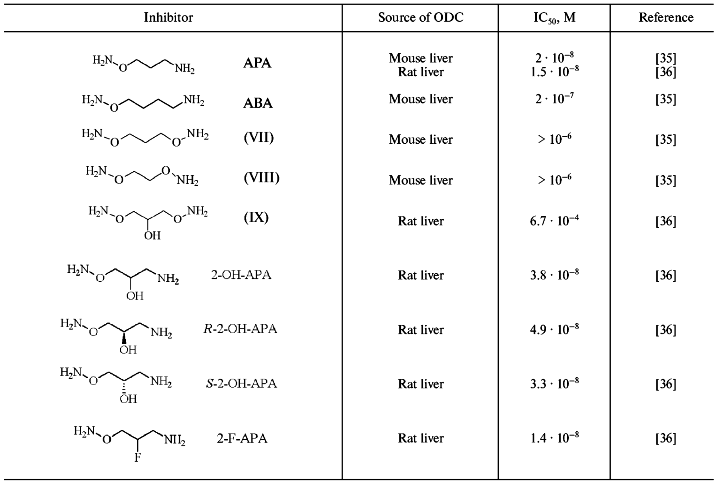
APA is an isosteric analog of Put but the introduction of H2NO-group into Put converted the latter into a carbonyl reagent. The positively charged amino group of APA plays anchoring functions and ensures proper binding of the inhibitor at the ODC active center. The aminooxy group (pKa ~ 5) of APA is unprotonated at physiological pH and responsible for formation of oxime with PLP in the enzyme active center. Inhibition of ODC did not develop in time, probably due to the activation of carbonyl group of PLP as the result of formation of internal aldimine with epsilon-amino group of Lys. Ornithine did not protect the enzyme from APA, and the activity of the inhibited ODC could not be restored upon addition of excess of the substrate. However, it was possible to obtain catalytically active enzyme after gel-filtration of E-I complex (elution buffer contained PLP). This observation was in a good agreement with the reversible character of PLP binding with apoenzyme (the affinity of PLP to apoenzyme is about 0.1-0.3 µM). The aminooxy analog of cadaverine (ABA) was 10-fold less active as compared with APA. Bis-aminooxy derivatives (VII-IX), which are non-protonated at physiological pH, did not exhibit any inhibitory effect even at 1 µM concentration. Specificity of action of APA was confirmed by low activity of the compound (IC50 about 0.1 mM) towards PLP-dependent ornithine transaminase and AdoMetDC.
Introduction of a hydroxyl group into the APA molecule (2-OH-APA) had practically no effect on the inhibitory potency of the analog; moreover, the activities of corresponding R- and S-isomers of 2-OH-APA were very similar (Table 3). On the contrary with APA, 2-OH-APA, and ABA, which were pseudo irreversible inhibitors of ODC (excess of the coenzyme was able to revert the inhibition), 2-F-APA was an irreversible inhibitor of ODC [37]. It is reasonable to assume that at the first stage 2-F-APA formed an oxime of ODC, while irreversible inhibition may come out either from alkylation of the essential for catalysis Cys360, or due to stabilization of E-I complex by additional strong hydrogen bond, which is quite typical for a fluorine atom. Unfortunately, the exact reasons for irreversible inhibition of ODC by 2-F-APA were not investigated in paper [37].
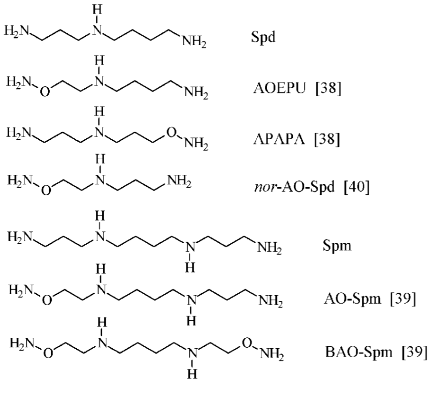
To conclude this section, it is necessary to remark that APA may be considered not only as a putrescine-like carbonyl reagent and respectively as an effective inhibitor of ODC, but also as a charge insufficient (H2NO-group has pKa ~ 5) putrescine analog. Since biological functions of polyamines are determined by superposition of both molecule structure and charge, an original set (Fig. 6) of charge insufficient aminooxy analogs of Spd [38] and Spm [39] were synthesized. These compounds turned out to be useful tools to investigate polyamine biochemistry. Aminooxy analogs of Spd and Spm turned out to be effectors of the enzymes of polyamine catabolism [41], they were found capable to penetrate inside cells and specifically inhibited the growth of tumor cells, exhibited low cytotoxicity, and regulated catabolic stability [42-45].
A general feature of all highly effective and specific hydroxylamine-containing inhibitors of AdoMetDC and ODC (see present paper), as well as GluDC [19], HisDC [18], GABA transaminase [20], and gamma-cystathionase [46] was the presence of properly positioned functional groups in alkyl fragment, that secured structural similarity with a substrate or with a product of the enzymatic reaction. These functional groups showed high affinity of the inhibitor to the enzyme active center at reverse stage, while aminooxy group formed an oxime with PLP or carbonyl-containing prosthetic group responsible for catalysis. Such an oxime of the enzymes may be considered as a mimetic of external aldimine--an intermediate compound common for all types of PLP-dependent transformations of amino acids. However, the above semi-empiric considerations require description in terms of structural similarity of E-I and E-S complexes. Since the correctness of this consideration may serve as a background of the algorithm for conversion of amino acid into its antagonist, a comparative X-ray investigation of complexes of aspartate aminotransferase (AspAT) with alpha-methylaspartate and aminooxyacetic acid was performed.Fig. 6. Structures of spermidine, spermine, and their aminooxy analogs.
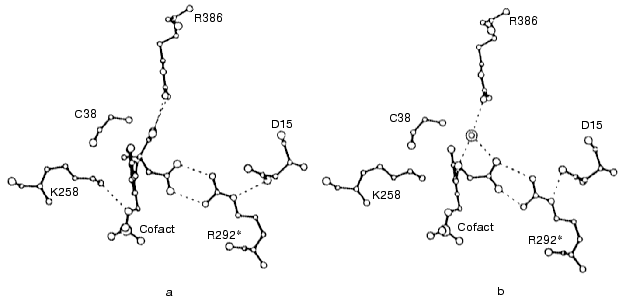
It turned out that in the complex of aminooxyacetic acid with mitochondrial AspAT (closed conformation), the HOOC-group of aminooxyacetate formed a hydrogen-bonded ion pair with Arg292 in a manner very similar to that of the beta-HOOC-group of alpha-methylaspartate and in both cases Arg292, in its turn, formed a salt bridge with Asp15 (Figs. 7a and 7b). Unlike the AspAT aldimine with alpha-methylaspartate, the oxime of the enzyme with aminooxyacetic acid lacks alpha-HOOC-group. In the case of alpha-methylaspartate this alpha-HOOC-group interacted with Arg386 of small domain (Fig. 7a). However, in AspAT oxime the place of alpha-HOOC-group was occupied with a water molecule, which was hydrogen bonded with the same Arg386 (Fig. 7b). Keeping in mind that formation of the oxime of mitochondrial AspAT with aminooxyacetic acid in closed crystal form produced only insignificant changes in cofactor orientation and conformation in the active site, as compared with corresponding complex formed by alpha-metylaspartate (Figs. 7a and 7b), it may be possible to consider structural similarity of E-I and E-S complexes to be confirmed and such an oxime of the enzyme does mimic corresponding external aldimine of the enzymatic reaction [47].
An important stage of the present study was the investigation of the interaction of hydroxylamine-containing inhibitors of AdoMetDC and ODC--AMA and APA, respectively--with cells. Both inhibitors were able to penetrate inside different cell types and to inhibit polyamine biosynthesis. Cell growth was reduced upon addition of polyamines in the media and this confirmed the specificity of the action of the inhibitors [48-51]. With the help of APA and AMA it was possible to confirm the key role of Spd in supporting growth and viability of cells [50, 51]; to demonstrate directly that AdoMetDC is synthesized as a proenzyme [52]; to investigate some peculiarities of the transport of polyamines and their analogs in cells [53]; to reveal that tumor cells with depleted polyamine poll are more sensitive to 5-F-Ura and methotrexate [51].Fig. 7. Schematic fragment of the structure of complexes of mitochondrial AspAT with alpha-methylaspartate (a) and aminooxyacetic acid (b) (taken from [47]).
Hence, from the result of the investigation of new possibilities of specific inhibition of AdoMetDC and ODC, key enzymes of polyamine biosynthesis, by substrate-like O-substituted hydroxylamines, it became possible to begin with an earlier unknown chemical compound that turned out to be an effective inhibitor of the enzyme, and to continue with specific regulation of cellular metabolism and growth.
The author's investigations of chemical regulation of polyamine metabolism were supported by the Russian Foundation for Basic Research grants 95-04-12278, 97-04-48708, and 00-04-48244.
REFERENCES
1.Marton, L. J., and Pegg, A. E. (1995) Ann. Rev.
Pharm. Tox., 35, 55-91.
2.Lee, Y. B., Park, M. H., and Folk, J. E. (1995)
J. Med. Chem., 38, 3053-3061.
3.Bazhenova, E. N., Deryabina, Yu. I., Eriksson, O.,
Zvyagilskaya, R. A., and Saris, N.-E. L. (1998) J. Biol. Chem.,
273, 4372-4377.
4.Reynolds, I. J. (1990) Adv. Pharm.,
21, 101-126.
5.Lopatin, A. N., Makhina, E. N., and Nichols, C. G.
(1994) Nature, 372, 366-369.
6.McCloskey, D. E., Yang, J., Woster, P. M.,
Davidson, N. E., and Casero, R. A. (1996) Clin. Cancer Res.,
2, 331-336.
7.Lindsay, G. S., and Wallace, H. M. (1999)
Biochem. J., 337, 83-87.
8.Kramer, D. L., Vujcic, S., Diegelman, P., Alderfer,
J., Miller, J. T., Black, J. D., Bergeron, R. J., and Porter, C. W.
(1999) Cancer Res., 59, 1278-1286.
9.McCann, P. P., and Pegg, A. E. (1992) Pharm.
Ther., 56, 195-215.
10.Pegg, A. E., and McCann, P. P. (1992) Pharm.
Ther., 56, 359-377.
11.Casero, R. A., and Woster, P. M. (2001) J.
Med. Chem., 44, 1-26.
12.Cohen, S. S. (1998) A Guide to the
Polyamines, Oxford University Press, New York.
13.Harik, S. I., and Snyder, S. H. (1973)
Biochim. Biophys. Acta, 327, 501-509.
14.Inoue, H., Kato, Y., Takigawa, M., Adachi, K.,
and Takeda, Y. (1975) J. Biochem. (Tokyo), 77,
879-893.
15.Wickner, R. B., Tabor, C. W., and Tabor, H.
(1970) J. Biol. Chem., 245, 2132-2139.
16.Rahiala, E. L., Kekomaki, M., Janne, J., Raina,
A., and Raiha, N. C. R. (1971) Biochim. Biophys. Acta,
227, 337-343.
17.Poso, H., Sinervirta, R., and Janne, J. (1975)
Biochem. J., 151, 67-73.
18.Reid, J. D., and Shepherd, D. M. (1963) Life
Sci., 2, 5-8.
19.Sastchenko, L. P., Severin, E. S., and Khomutov,
R. M. (1968) Biokhimiya, 33, 142-147.
20.Baxter, C. F., and Roberts, E. (1961) J. Biol.
Chem., 236, 3287-3294.
21.Khomutov, A. R., and Khomutov, R. M. (1986)
Bioorg. Khim., 12, 1662-1674.
22.Khomutov, R. M., Zavalova, L. L., Sirku, V. I.,
Artamonova, E. Yu., and Khomutov, A. R. (1983) Bioorg. Khim.,
9, 130-131.
23.Artamonova, E. Yu., Khomutov, A. R., Zavalova, L.
L., and Khomutov, R. M. (1986) Bioorg. Khim., 12,
206-212.
24.Pegg, A. E. (1984) Cell Bochem. Funct.,
2, 11-15.
25.Wetkamp, E. L. C., Dixon, H. B. F., Khomutov, A.
R., and Khomutov, R. M. (1991) Biochem. J., 277,
643-645.
26.Kolb, M., and Barth, J. (1985) Liebigs
Ann. Chem., 1035-1040.
27.Pegg, A. E., Jones, D. B., and Secrist III, J. A.
(1988) Biochemistry, 27, 1408-1415.
28.Pankaskie, M., and Abdel-Monem, M. M. (1980)
J. Med. Chem., 23, 121-127.
29.Anton, D. L., and Kutney, R. (1987)
Biochemistry, 26, 6444-6447.
30.Diaz, E., and Anton, D. L. (1991)
Biochemistry, 30, 4078-4081.
31.Li, Y.-F., Hess, S., Pannell, L. K., Tabor, C.
W., and Tabor, H. (2001) Proc. Natl. Acad. Sci. USA, 98,
10578-10583.
32.Parsons, I. D., Persson, B., Mekhalfia, A.,
Blackburn, G. M., and Stockley, P. G. (1995) Nucleic Acids Res.,
23, 211-216.
33.Mecalf, B. W., Bey, P., Danzin, C., Jung, M.,
Casara, P., and Vevert, J. P. (1978) J. Am. Chem. Soc.,
100, 2551-2553.
34.Pegg, A. E., Shantz, L. M., and Coleman, C. S.
(1995) J. Cell. Biochem., 22, 132-138.
35.Khomutov, R. M., Denisova, G. F., Khomutov, A.
R., Belostotskaya, K. M., Shlosman, R. B., and Artamonova, E. Yu.
(1985) Bioorg. Khim., 11, 1574-1576.
36.Stanek, J., Frei, J., Mett, H., Schneider, P.,
and Regenass, U. (1992) J. Med. Chem., 35, 1339-1344.
37.Mett, H., Stanek, J., Lopez-Ballester, J. A.,
Janne, J., Alhonen, L., Sinervirta, R., Frei, J., and Regenass, U.
(1993) Cancer Chemother. Pharm., 32, 39-45.
38.Khomutov, A. R., and Khomutov, R. M. (1989)
Bioorg. Khim., 15, 698-703.
39.Khomutov, A. R., Vepsalainen, J., Shvetsov, A.
S., Hyvonen, T., Keinanen, T. A., Pustobaev, V. N., Eloranta, T. O.,
and Khomutov, R. M. (1996) Tetrahedron, 52,
13751-13766.
40.Khomutov, A. R., Shvetsov, A. S., Vepsalainen, J.
J., and Kritzyn, A. M. (2001) Tet. Lett., 42,
2887-2889.
41.Eloranta, T. O., Khomutov, A. R., Khomutov, R.
M., and Hyvonen, T. (1990) J. Biochem. (Tokyo),108,
593-598.
42.Hyvonen, T., Keinanen, T. A., Khomutov, A. R.,
Khomutov, R. M., and Eloranta T. O. (1992) J. Chromatogr.,
574, 17-21.
43.Hyvonen, T., Keinanen, T. A., Khomutov, A. R.,
Khomutov, R. M., and Eloranta, T. O. (1995) Life Sci.,
56, 349-360.
44.Khomutov, A. R., Schvetsov, A. S., Vepsalainen,
J., Kramer, D. L., Hyvonen, T., Keinanen, T. A., Eloranta, T. O.,
Porter, C. W., and Khomutov, R. M. (1996) Bioorg. Khim.,
22, 557-559.
45.Turchanowa, L., Schvetsov, A. S., Demin, A. V.,
Khomutov, A. R., Wallace, H. M., Stein, J., and Milovic, V. (2002)
Biochem. Pharm., 64, 649-655.
46.Gabibov, A. G., Shhuster, A. M., Khomutov, A. R.,
Kostina, M. B., Khirs, E. N., Goryachenkova, E. V., and Khomutov, R. M.
(1996) Dokl. RAN, 350, 405-407.
47.Markovic-Housley, Z., Schirmer, T., Hohenester,
E., Khomutov, A. R., Khomutov, R. M., Karpeisky, M. Ya., Sandmeier, E.,
Christen, Ph., and Jansonius, J. N. (1996) Eur. J. Biochem.,
236, 1025-1032.
48.Hyvonen, T., Alakuijala, L., Andersson, L.,
Khomutov, A. R., Khomutov, R. M., and Eloranta, T. O. (1988) J.
Biol. Chem., 263, 11138-11144.
49.Persson, L., Khomutov, A. R., and Khomutov, R. M.
(1989) Biochem. J., 257, 929-931.
50.Kramer, D. L., Khomutov, R. M., Bukin, Yu. V.,
Khomutov, A. R., and Porter, C. W. (1989) Biochem. J.,
259, 325-331.
51.Milovic, V., Turchanova, L., Khomutov, A. R.,
Khomutov, R. M., Caspary, W. F., and Stein, J. (2001) Biochem.
Pharm., 61, 199-206.
52.Antelli, R., Stjernborg, L., Khomutov, A. R.,
Khomutov, R. M., and Persson, L. (1991) Eur. J. Biochem.,
196, 551-556.
53.Kramer, D. L., Miller, J. M., Bergeron, R. J.,
Khomutov, R. M., Khomutov, A. R., and Porter, C. W. (1993) J. Cell.
Physiol., 155, 399-407.