REVIEW: Proteomic Technologies in Modern Biomedical Science
V. M. Govorun1,2* and A. I. Archakov1
1Orekhovich Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, Pogodinskaya ul. 10, Moscow, 119992 Russia; fax: (095) 245-0857; E-mail: inst@ibmh.msk.su2Institute of Physico-Chemical Medicine, Ministry of Health of Russian Federation, ul. Malaya Pirogovskaya 1a, Moscow, 119992 Russia; E-mail: govorun@hotmail.ru
* To whom correspondence should be addressed.
Received April 17, 2002; Revision received May 25, 2002
This review highlights modern technologies employed in proteomics. Methods of sample preparations are discussed with special emphasis on the requirements for preparation of biological material, which may seriously influence the results of proteomic studies. Methods of solubilization, electrophoresis, chromatographic protein separation, and visualization of protein spots in gels are described. Modern methods of mass spectrometry used in proteomic studies include combination of protein chips with mass spectrometry. The review also describes approaches of functional proteomics, i.e., interactomics, and also bioinformatic resources used in proteomics for image analysis of 2D-gel-electrophoresis and for identification of protein sequences by mass spectra.
KEY WORDS: proteomics, biotechnology, electrophoresis, chromatography, protein chip
Abbreviations: AD) transcription activator domain; ASB) alpha-sulfobetaine; BD) DNA-binding domain; BIA) biomolecular interactive analysis; CAI) codon adaptation index; CBI) codon bias index; CHAPS) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid; 2D-electrophoresis) two-dimensional electrophoresis; DTE) dithioerythritol; DTT) dithiothreitol; ELISA) enzyme-linked immunosorbent assay; ESI) electrospray ionization; ESTdb) Expressed Sequence Tags database; HUPO) Human Proteome Organization; HUGO) Human Genome Organization; ICAT) isotope coded affinity tags; IEF) isoelectrofocusing; IPG) immobilized pH gradient; LC) liquid chromatography; LCM) laser capture microdissection; MALDI) matrix assisted laser desorption ionization; MS) mass-spectrometry; MOWSE) molecular weights search; NCBInr) National Center for Biotechnology Information; NP-40) Nonidet P-40; PIR) protein identification resource; PISA) protein in situ array; SELDI) surface enhanced laser desorption ionization; TBP) tributylphosphine; TOF) time of flight.
The term “proteomics” or “proteome” was introduced
in 1995 [1]. However, as a notion it appeared in
the modern biology thirteen years earlier when Anderson and Anderson
proposed the creation of a molecular atlas of human proteins [2]. The major goal of proteomics as a science is
making inventory of all proteins encoded in the genome of a certain
organism and analysis of interaction of these proteins [3, 4]. Methodologically,
proteomics is based on highly efficient methods of separation and
analysis of proteins in living systems. Ideally, the use of these
methods will provide exhaustive information on biochemical properties
of proteins in living systems (level of protein expression,
posttranslational modifications, protein-protein interactions, etc.).
Now this science is intensively developed in many countries. Proteomics
takes leading positions in research programs of modern applied and
basic biology, pharmaceutics, and related areas. In 2001 an
international consortium of scientists, businessmen, and politicians
founded a new organization, the Human Proteome Organization (HUPO). Its
major goal is making an inventory of all human proteins, creation of
molecular protein atlases of cells, organs, tissues, schemes of
protein-protein interactions, development of special informational
databases, and searching for specific markers of pathological
processes. In contrast to a previous project, HUGO (Human Genome
Organization), founded for complete nucleotide sequencing of human DNA,
HUPO did not announce precise aims and dates for termination of these
projects because it is nearly impossible to predict the exact number of
individual proteins in cells under normal conditions and especially in
various diseases.
What does term “proteome” (or proteomics) mean? Which methodological approaches does this science employ? What are the main achievements of this science and what are the main trends in technologies of proteomic analysis in the coming years? In reality it is difficult to give correct comprehensive answers to all these questions because of very high rates of development of technological and methodological bases of proteomics. Every two years leading suppliers update their equipment (mass spectrometers, chromatographs, etc.). However, in contrast to many other biological sciences of postgenomic era proteomics represents an almost unlimited area of our knowledge because of almost countless number of research objects (proteins). According to calculations made by various experts the exact numbers of proteins varies in the range from 1*106 to 5*106. So, at the moment full inventory of all proteins is almost impossible [5]. However, it should be noted that protein inventory is only one of the major goals of proteomic analysis. We should learn more about principles of protein-protein interactions, regulation of their concerted functioning, and posttranslational modifications. In contrast to other sciences proteomic organizations can be roughly subdivided into two principally distinct types. Several decades of large proteomic companies with the budget exceeding one billion US dollars represent one type of proteomic organizations (Table 1). Numerous proteomic service firms in universities and research institutions and proteomic academic groups which are characterized by extensive experience but limited technical and financial resources form the second type of the proteomic organizations [6]. It is clear that technologies used by large proteomic companies and university centers differ by various parameters (price, productivity, aims and objectives, etc.). In this review we consider the main “classic” technologies which are often used in small proteomic centers which may be successfully realized in Russia.
Table 1. Leading proteomic companies

Note: 2DE/MS is two dimensional electrophoresis and mass
spectrometry.
METHODS OF SAMPLING, SOLUBILIZATION, AND SEPARATION OF PROTEINS
FOR PROTEOMIC ANALYSIS
Usually, traditional handbooks on proteomic analysis pay little (if any) attention to so-called sampling systems because they represent an intrinsic part of technical skill of laboratory or clinical personnel. However, one year functioning of the Center of Proteomic Studies at Orekhovich Institute of Biomedical Chemistry revealed that this is an important problem and there are some objective factors which seriously deteriorate protein separation. Consequently, correct interpretation of results becomes nearly impossible. Below we describe the main requirements for the sampling of biological material that significantly influence the quality of proteomic studies.
1. Presence of Single Type of Cells Which Are Synchronized by Cell Cycle and Stage of Differentiation
In the case of laboratory strains of eu- and prokaryotic microorganisms the employment of traditional technologies for cell cultivation [7] provide required synchronization and differentiation. Using strains of E. coli and Bacillus subtilis cells it was shown that the proteome map (protein profile after 2D-electrophoretic separation) is significantly influenced by growth phase, cultivation conditions, the strain employed, etc. [8-10]. Clinical isolates of some bacterial strains require special cultivation and it is difficult to control and standardize parameters of their growth in vitro. In these cases proteomic mapping requires additional experiments using such technologies as biochips, allowing the determination of the level of gene expression, etc. [11].
Preparation of cells of multicellular organisms which would be characterized by the same stage of differentiation represents a difficult task. As a rule, data on mRNA and protein expression levels obtained using cell culture and directly from tissues and organs significantly differ. [12]. Unfortunately, materials of biopsy and autopsy often represent a mixture of various cell and tissue types (e.g., connective tissue, vascular endothelium, organ-specific cells, blood cells, etc.). This seriously complicates interpretation of results. Recently, laser microdissection technology has been employed in post-genomic studies, particularly in proteomics, for isolation of homogenous cell populations from heterogeneous tissue biopsy materials [13-15]. Laser capture microdissection (LCM) is a method, which allows separation of the same morphological type cells from tissue samples of microscopic preparations. This method is effective for isolation of cells representing less than 5% in the analyzed sample irrespectively to morphological properties of the extracellular matrix.
This method was originally developed in 1996 by Emmert-Buck et al. [16]. In 1997 this method was standardized and developed as a commercial automated system by Arcturus Engineering Inc. (USA). The equipment for LCM includes a light microscope equipped with a source of laser emission and a computer with software for control of analytic stages and treatment of data obtained. This analysis requires standard microscopic preparations using ordinary microscopic optics for routine studies. The principle of the laser microdissection consists in laser-beam activation of special transfer film located just above the analyzed sample, fixation of this material on this film followed by transfer of the film with fixed material and its use in subsequent studies.
The transfer film positioned above the sample is transparent and this allows visual evaluation of the area of interest in the microscopic preparation. In the commercial systems of Arcturus Engineering Inc. the transfer film is fixed at the lower side of exchangeable transparent capsules and this significantly facilitates film positioning and subsequent sample transfer.
Unfortunately, these devices have limited abilities and microdissection procedures usually give from 25,000 to 100,000 cells [17]. This influences the strategy of proteomic analysis. The sensitivity of mass-spectrometric protein detection (products of trypsinolysis) should be at least 0.1-1.0 fmol and in some cases the amount of protein is below these limits. However, subsequent development of this approach and improvement of LCM-equipment is very promising for in vivo systemic proteomic studies for creation of proteomic atlases of cells, tissues, and organs. Perhaps, in the future such studies will displace studies employing heterogeneous preparations and cell cultures as inadequate experimental models.
2. Methods of Protein Solubilization
The choice of methods of protein solubilization for proteomic mapping depends on the technology used for subsequent separation of proteins.
According to the classical scheme of proteomic analysis the primary separation of proteins by means of 2D-electrophoresis implies the employment of high concentration of a chaotropic agent (urea) and detergents. Sometimes the employment of several related chaotropic agents (e.g., urea and thiourea) increase effectiveness of solubilization. Table 2 summarizes the most frequent combinations of reagents for protein solubilization.
Table 2. Combination of reagents used for
protein solubilization
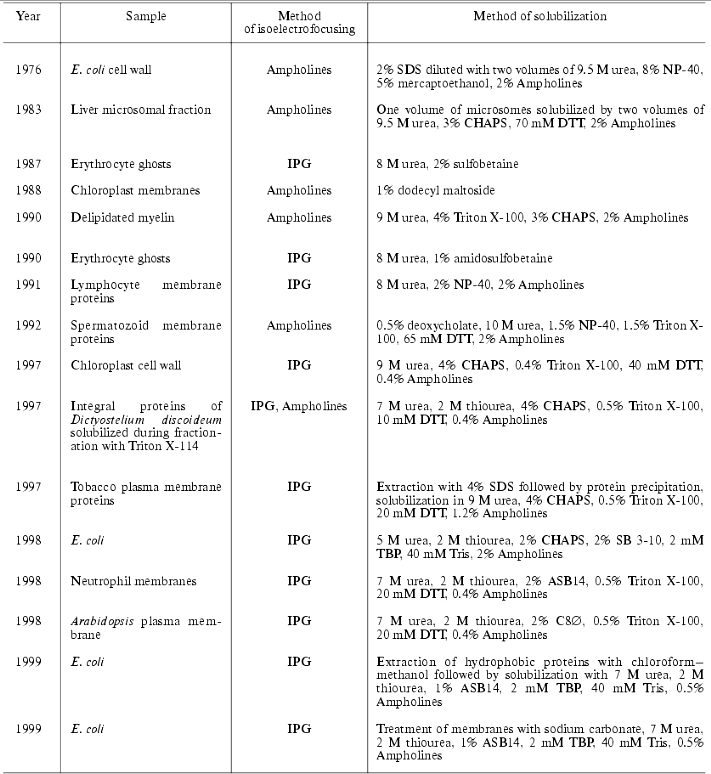
It should be noted that now the standard solubilization scheme involves the use of 8 M urea, 4% CHAPS (3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonic acid), 0.1-0.2% Ampholines, 3-10 and 10-100 mM DTT (dithiothreitol) or DTE (dithioerythritol). Most suppliers of immobilized pH gradients (IPG, Immobilines) recommend this procedure for the first dimension of electrophoretic separation (isoelectrofocusing, IEF). Usually, high concentrations of DTT/DTE are used, but these compounds are weak acids and the process of sulfhydryl group reduction is reversible. During IEF procedure these compounds migrate to the anode; the latter results in the decrease of their actual concentrations, which may cause reoxidation of sulfhydryl groups and formation of horizontal lines after visualization of separated products. Such lines are frequent artifact seen during 2D-electrophoresis [18]. The use of mercaptoethanol is not recommended now.
Phosphines, particularly tributylphosphine (TBP), are an alternative to the use of DTT/DTE. Their use allows decreasing the concentration of the reducing agent to 2 mM. The use of TBP significantly increased solubilization of keratin proteins, which are characterized by a large number of sulfhydryl groups [19]. Since TBP lacks any charge it has certain advantages when alkaline gradients are used. The use of phosphines allows reduction and alkylation of proteins after their separation by IEF. In contrast to DTT, TBP does not interact with some alkylating reagents such as acrylamide and 4-vinylpyridine [20]. However, such alkylating agents as iodoacetamide and its derivatives cannot be used together with TBP and as in the case of DTT/DTE use this requires a two-step procedure [21]. Other variations of protein solubilization also exist and their use for protein separation in 2D-electrophoresis depends on properties of analyzed biological material, specific goals, and laboratory equipment [22-24].
At the end of this section on protein solubilization for 2D-electrophoresis let us consider several examples illustrating successful use of 2D-electrophoresis for fractionation of membrane proteins. The latter is a rather important task because the standard procedure of protein solubilization for IEF (see above) is preferentially employed for separation of water soluble proteins. Most proteomic maps contain information mainly about water soluble proteins, whereas membrane proteins possessing very important functions in the cell have been analyzed just in relatively few proteomic studies.
It should be noted that commonly used detergents (CHAPS, NP-40, or Triton X-100) in combination with urea are sufficient for successful identification of some membrane (but not integral) proteins (Table 3). During last three years sulfobetaine derivatives are successfully used for solubilization of membrane proteins. (CHAPS also belongs to this group.) The most interesting detergents used for fractionation of membrane proteins followed by 2D-electrophoretic separation are ASB14 and C8. containing 14 alkyls and p-phenyloctyl, respectively. Both detergents in combination with 7 M urea and 2.5 M thiourea were successfully used for solubilization of integral membrane proteins of E. coli and Arabidopsis thaliana [25, 26]. Additional procedure promoting successful fractionation of integral membrane proteins includes primary treatment of membrane proteins in initial membrane preparations with the alkaline solution containing 50-100 mM sodium carbonate, pH 11, followed by subsequent solubilization of the membrane preparation using one of above mentioned methods [26]. Sometimes alkaline carbonate lysis is directly used for solubilization of membrane proteins for IEF [27].
Table 3. Identification of membrane
proteins

The last three years are characterized by gradual change in the strategy of protein fractionation during the first stage of proteomic studies and the development of new methods of multimer chromatography in combination with tandem mass spectrometry. The first use of 2D-chromatography was reported by Link et al. [33]. In contrast to 2D-electrophoresis chromatographic technologies significantly extend the list of organic solvents, detergents, and surfactants used for protein solubilization. A combination of solubilization and isotope labeling of peptides obtained by trypsinolysis or cyanogen bromide hydrolysis of proteins significantly increased effectiveness of proteome mapping including integral membrane proteins; however, this is a more expensive and time-consuming procedure than 2D-electrophoresis [34].
3. Protein Separation
2D-electrophoresis. Here we will not describe in detail the procedure of 2D-electrophoresis introduced in the practice of biochemical laboratories by O'Farrel in 1975 [35]. Such information has been published elsewhere [36-42]. It is also available on the following Internet-sites: http://www.weihenstephan.de/blm/deg/manual/manualwork2html02testp6.htm and http://www.expasy.ch/ch2d/protocols. Here we consider the main methodical improvements of this technology, which significantly increased analytical capacities and reproducibility of results. The majority of the improvements are related to the first dimension of protein fractionation, isoelectrofocusing.
Polyacrylamide gel-immobilized pH gradients (IPG) for isoelectrofocusing. Initially the use of IPG was described by Bjellqvist et al. in 1982 [43]. The main protocol for the use of IPG for 2D-electrophoresis was published by Görg et al. in 1988 [40]. During the next fourteen years this method became commonly accepted. It effectively displaced Ampholines as the source for pH gradient for the following reasons:
a) isoelectrofocusing of proteins is not accompanied by so-called “cathode shift” [40];
b) strips with IPG of various shape (linear, exponential, etc.) are commercially available; they are ready for use after a rehydration procedure. Procedures for laboratory preparation of IPG are also available [44];
c) strips with Immobilines have greater protein loading capacities than polyacrylamide carriers containing ampholines [45];
d) suppliers produce strips with IPG at alkaline region up to pH 12.5. This allows effective fractionation of basic proteins (however, in this case expiry date is limited to one month from the date of IPG issue);
e) strips with narrow (1-2) or ultra-narrow (0.2) ranges of pH-gradients are commercially available. This significantly increases their loading for isoelectrofocusing (up to 4-10 mg of protein versus 0.1-1.0 mg of protein in the case of ordinary strips with the range of pH 3-10 or 5-8). Using such strips it is possible to identify proteins which are present in biological material in rather low quantity (10-100 copies per cell) [46]. Aebersold and his colleagues also demonstrated that use of narrow (1.0) pH gradients for separation of yeast proteins allowed identification (by using tandem mass spectrometry) of proteins encoded by genes possessing Codon Bias Index (CBI) or Codon Adaptation Index (CAI) within 0.2-0.1 [47]. This increases the total number of protein spots on a synthetic gel up to 10,000 versus 1,000-2,500 obtained by using standard pH gradients (3-10, 5-8).
Various methods of fractionation of protein extracts before isoelectrofocusing have been used to increase the resolution of the IEF. For example, in 2000 Herbert and Righetti described a special multi-chamber apparatus for an additional step of preparative IEF combined with separation of protein mixtures into a subfraction that is used before analytical IEF [48]. Protein separation in this so-called multi-chamber electrolyzer is based on “capture” of fractionated proteins by membranes containing Immobilines with certain pH values. Introduction of such procedure before analytical IEF effectively removes amines, fatty acids, salts (and other components of biological extracts influencing results of IEF) from the analyzed protein mixture [49]. Suppliers of equipment and reagents for proteomic studies produce kits for effective extraction of proteins and removal of major proteins (e.g., plasma proteins), lipid, salts and other contaminants influencing IEF process.
Methods for visualization of protein spots after separation by 2D-electrophoresis. All standard procedures of protein staining are used in proteomic studies. However, some of them are not applicable for mass-spectrometry methods of protein identification due to irreversible modification of amino acids. Photochemical methods of staining with silver [50, 51], Coomassie colloid solution [52], and a special fluorescent dye Sypro Ruby [53] are the most popular and widely used methods of staining. Applicability of certain methods of staining is usually determined by the possibility of quantitative or semi-quantitative evaluation of protein expression during comparison of two or more samples. This is a very important precondition, because the main methods of mass spectrometry, used for identification of proteins or tryptic fragments extracted from the protein spot are qualitative rather than quantitative methods. If 2D-electrophoresis is used for protein separation it is necessary to select staining that gives quantitative or semiquantitative results. Photochemical silver staining is not a quantitative method. Alternative procedure of silver staining in combination with glutaraldehyde is a semiquantitative technology. However, the latter is not applicable in the case of mass spectrometry methods of peptide identification due to irreversible modification of lysine and arginine residues; this makes impossible trypsinolysis [51]. Approaches used in microarray transcriptome studies are intensively advertised for proteomics [54]. Pharmacia-Amersham-Biotech (USA) offers special kits for labeling of protein extract with two fluorophores, Cy3 and Cy5, exhibiting distinct fluorescent spectra. This allows separating protein extracts in one gel and evaluating relative quantitative changes in protein content by using program algorithms of a differential display. Protein separation is still of the same type but application of these kits avoids additional electrophoretic experiments [55]. This approach was successfully used for determination of differential levels of mRNA. It is possible that this approach will be also used in proteomic studies employing protein separation by means of 2D-electrophoresis.
Multidimensional chromatography. This is a group of chromatographic methods using various sorbents but one eluent system for chromatographic separation of protein fragments obtained after trypsinolysis or cyanogen bromide hydrolysis. In most cases a carrier consists of two sorbents, strong cation exchanger (SCX) and a reversed phase (RP-18) [56, 57]. Chromatographic methods and equipment used in proteomic studies are combined (in online mode) with electrospray mass-spectrometry detector; they have eluent flow rate of 1-100 nl/min. This increases not only resolution capacity of chromatographic separation of peptides but also increases sensitivity of mass-spectrometry protein determination both in nanoESI-MS and MS-MS modes [58]. Stepwise gradient elution of proteins (peptides), which uses special valves for eluent switching is another example of multidimensional chromatography [59]. There is a special example of multidimensional chromatography implemented by the Ciphergen companythat includes a kit of metallized strips with various chromatographic phases and special protein ionization technology without additional enzymatic treatment. We consider this kind of multidimensional chromatography in the section on protein arrays.
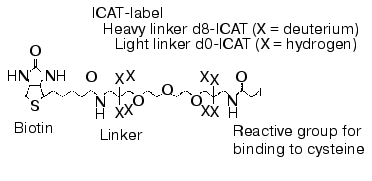
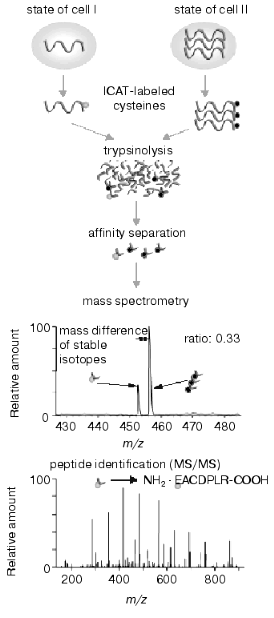
The intensive development of multidimensional chromatography methods and their applications may be due to the following reasons. First, in spite of constant use and continuous improvement of 2D-electrophoresis, this method still remains some kind of art rather than routine technique. It cannot be automated and in spite of high resolution capacity it is characterized by lower productivity than other methods used for protein separation. Second, new methods based on selective isotope labeling of peptides appeared and these methods are constantly improved. These methods effectively solve the problem of quantitative comparison of control and experimental samples. Using automated mode and repeated chromatographic cycles on bi- and triphasic sorbents with subsequent on-line mass spectrometry analysis and identification of peptides these methods are also effective in separation and identification of hydrophobic proteins. Isotope coded affinity tags (ICAT) method initially introduced by Aebersold and his colleagues [60] is one of these methods. Commercially available kits using ICAT-technology are distributed by Applera (USA). The principle of this method consists of separate trypsinolysis of protein extracts from control and experimental samples followed by subsequent administration of a special label (Fig. 1), interacting with cysteine residues. This label contains a reactive group for covalent binding, a linker containing eight atoms of either hydrogen or deuterium (representing its own isotope code) and biotin. The latter is an affinity tag for extraction from protein mixture on affinity columns. Purified mixture of cysteine-labeled peptides from control and experimental samples is pooled and separated using multidimensional chromatography with nano-ESI-MS or MS-MS detection with subsequent quantification and identification of peptides of the same type. Due to isotope differences of the cysteine reagent linker used for peptide labeling of control and experimental samples molecular masses of the same peptides from these samples differ by 8 daltons. Figure 2 gives a schematic presentation of this technology.
Fig. 1. Structural formula of ICAT-label.
The authors of this technology illustrated its functioning by analyzing yeast protein extract. Theoretically, the yeast proteome map consists of 6000 proteins (genome capacity) and calculated number of fragments (peptides) obtained after trypsinolysis is about 300,000. However, only 30,000 peptides contain cysteine residues and they may be isolated by means of ICAT-technology. Taking into consideration complete nucleotide sequence of the yeast genome about 92% of proteins contain at least one cysteine and, consequently, ICAT-technology can analyze 5500 proteins. However, in reality it is not possible to reach this calculated value because of limitations of separating systems, the existence of modified groups of amino acids, and some technical problems (such as rates of mass spectrometry, resolution capacity of mass-spectrometers, technical capacities of chromatographs, program algorithms of peak identification). At the moment effectiveness of ICAT and related technologies is comparable with that of methods of classic proteomics in number of identified proteins. In the case of E. coli proteins the number of identified proteins is about 70%, whereas in the case of eukaryotes this value does not exceed 20-30%. Nevertheless, it is clear that automated chromatographic systems with on-line operated mass-detectors have evident advantages for the future both by productivity of analysis and analytic capacities. In contrast to 2D-electrophoresis, ICAT-technology can be applied and has successfully been applied for differential comparison of integral membrane proteins [61].Fig. 2. Scheme of ICAT-technology.
MASS SPECTROMETRY
Historically the term “proteomics” appeared after introduction of new methods in mass-spectrometry allowing ionization of biological macromolecules without their chemical destruction. There are special handbooks and excellent reviews on methods of mass-spectrometry and their application in proteomic studies [62-75] and so we leave out of consideration detailed characteristics of this analytic method.
There are two main modifications of ionization of native proteins and peptides: matrix-assisted laser desorption-ionization (MALDI) and electrospray ionization (ESI). Time of flight (TOF) mass-spectrometers are mainly used for primary spectra of peptides up to 35-40 kD. MALDI-TOF-mass-spectrometers are used for protein identification by means of peptide fingerprinting (by treatment of the sample with trypsin, cyanogen bromide, or any other endopeptidase, peptide restrictase). They are suitable for analysis of material obtained from organisms with known complete nucleotide sequence of genome. Tandem mass-spectrometers are equipped with ion trap, quadrupole chamber for fragmentation or hybrid quadrupole-time of flight (Q-TOF) apparatus. They are used for protein sequencing during mass-spectrometry of products of fragmentation of primary molecular ions of peptides or proteins within the format of chromatography-tandem mass-spectrometry (LC-MS/MS or nanospray using the principle of ESI). Let us consider principles of operation of these mass-spectrometers and the latest technical achievements, which overcome some traditional limitations existing in protein mass-spectrometry.
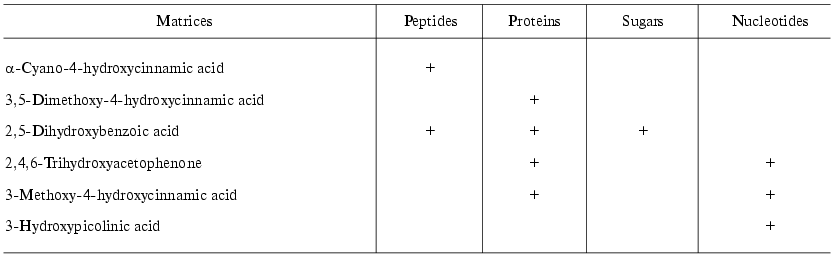
Matrix-assisted laser desorption-ionization (MALDI). In the process of preparation the analyzed sample is mixed with a special matrix which is a proton donor. This matrix interacts with proteins and forms crystalloid structures with them. These structures are targets for the laser emission; after a short laser impulse (2-5 nsec) crystalloids formed by the organic matrix and protein are evaporated. Such mode of ionization on the surface of a chip (in the presence of the organic matrix) results in formation of single charged protein ions, although protein ions of two or three charges can also be formed. Industrial MALDI-TOF mass-spectrometers are equipped with UV- (336 nm) or IR-lasers as the emission source. The main types of matrices are summarized in Table 4. Modern MALDI-TOF mass spectrometers may also be equipped with a reflectron, which increases resolution of the spectrometer (Fig. 3). MALDI-TOF spectrometers are also equipped with special post-source decay apparatus for isolation of peptide ions and their subsequent fragmentation and microsequencing. The latest MALDI-TOF mass-spectrometers are hybrid double TOF-TOF spectrometers which are able to get information on sequence of analyzed peptides. Since 2001-2002 these spectrometers are simultaneously produced by Applera and Bruker Daltonics (USA). Spectrometers produced by Applera employ ion trap for peptide isolation after the first cycle of analysis; the isolated peptide is subjected to subsequent fragmentation and separation. The spectrometer TOF-TOF Ultraflex produced by Bruker Daltonics employs special ion lift, which also releases molecular ions for their subsequent fragmentation and analysis. Although these spectrometers are quite expensive they will be used in protein mass spectrometry because they can produce secondary single charged ions of fragmentation products, which facilitates peptide sequencing.
Table 4. Main types of matrices for MALDI
with UV-laser
Electrospray ionization (ESI). The use of ESI results in dispersion of the analyte solution which is supplied via a thin capillary under high voltage (up to 20 kV). Big electrostatic charge causes flow brakes followed by subsequent dispersion into microdrops. Charged microdrops are readily dried and their charges ionize the analyte. Thus, in contrast to MALDI the employment of ESI results in formation of multicharged ions. In the case of ESI the sample flow rate varies in a wide range (1-500 µl/min) and so direct connection with chromatographic systems without flow separation becomes possible. However, this requires high sample consumption (10-5-10-8 M). Modern mass-spectrometers used for analytical proteomic studies usually contain special equipment, so-called nanoelectrospray (nanoESI) allowing sample administration at the flow rate of 0.01-0.5 µl/min (using the same sample concentrations). This makes possible direct connection of ESI mass-spectrometer detectors with multidimensional chromatography. This direction promotes further development of proteomic studies excluding 2D-electrophoresis.Fig. 3. Principle of MALDI-TOF mass-spectrometer operation.
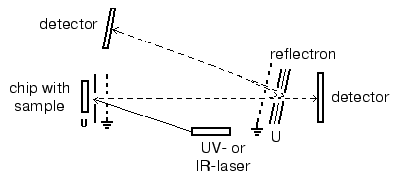
The reversed dependence between the intensity of signal and mass of the analyzed protein is one of the major limitations for protein determination by mass-spectrometry. It is generally accepted that TOF mass-spectrometers produce reliable determination of m/z ratio for protein within the range of molecular masses from 500 to 50,000 daltons. In 2001 Twerenbold et al. described a molecular cryodetector for TOF mass-spectrometers [76]. This cryodetector is a microcalorimeter, which can operate in a wide range of masses of the analyzed proteins and polymers (polyethylene glycol) without reduction in sensitivity.
Protein chips. Protein chips produced by Ciphergen are the most known and commercially successful example of protein chip production. This company developed an apparatus combining chromatographic protein chips with TOF mass spectrometer detector (ProteinChip SystemTM). The general purpose of the system is to discover biomarkers of pathological conditions in complex biological media.
Each metallic array contains eight well-chips with tightly immobilized chromatographic sorbent (hydrophilic phase (SiO2), hydrophobic phase (C18), weak cation exchanger, strong cation exchanger, metal-affinity carrier). Chips with activated surface for binding of any affinity sorbent (selected by user) are also commercially available.
Chip strips are fixed into a structure similar to a 96-well plate and then pairs of control and experimental samples are applied on each row of chromatographic sorbents. Each set of chips (experiment versus control) is washed in a certain way. After washing a solution of photosensitive compound (matrix) which is crystallized when drying is added to chips. The substances are evaporated from the chip surface and ionized into a TOF mass-spectrometer detector using laser emission. In the case of positive results there are differences in control and experimental (norm versus pathology) mass spectra on a chip with one of a few sorbents and using certain mode of washing. At the first stage it is impossible to detect which particular substance is a marker of a particular pathology. It is only possible to evaluate roughly molecular mass of this substance. Subsequently, the chip containing such component may be treated with protease and resultant peptide fingerprint may be used for identification of this biological marker.
This technology including direct TOF mass spectrometry from an active chromatographic surface is known as SELDI (surface-enhanced laser desorption-ionization). For convenient interpretation of results, Ciphergen also supplies special software, which allows to present results of mass-spectrometry determination of biological material retained on the chromatographic sorbent in the form of densitograms with indication of the intensity of each “band” (peak of molecular protein ions analyzed by mass spectrometry). Thus, in contrast to qualitative evaluation of the peak of the ionized peptides which is traditionally used in all mass spectrometry methods, Ciphergen developed equipment allowing relatively quantitative mass-spectrometry evaluation of proteins to be detected. The use of SELDI technology significantly extended the limits of analyzed protein mass up to 250 kD. SELDI thus can be used in medical centers without special mass spectrometry training of personnel. There are several reports on the discovery of new markers of pathological processes, e.g., tumors of various localization using SELDI technology [77, 78].
Besides this variant of protein chips representing an example of successful application of multidimensional chromatography, other devices that replace expensive multistage procedures of protein analysis have also been developed. Some of them can be employed in clinical practice.
The first group includes miniaturized devices employing (by analogy with DNA-chips) monoclonal antibodies or their analogs (aptamers, AffibodyTM, single chain antibodies, etc.) for the development of microarrays on known markers, which are now determined by traditional methods of laboratory diagnostics. Chemistry of protein immobilization and devices, producing spatially coordinated reagent distribution are similar to those used for preparation of DNA-chips. Glaucus Proteomics (Holland) is one of the unquestioned leaders in this field. This company produces monoclonal antibodies against all expressed human proteins followed by their application onto a solid phase and micro-ELISA analysis. Protein chips for determination of cytokines, alkaline phosphatases, and acute phase proteins are now commercially available. In 2003-2004 whole panels of protein chips for quantitative determination of several hundred of clinically important proteins will become available.
The second group includes technologies for evaluation of protein-protein interactions using in vitro translation followed by product application onto a solid phase. For example, PISA (Protein In Situ Array) technology employs PCR-products and cell-free translation systems for expression of proteins. This approach is successfully used in studies of protein-protein interactions [79]. Using cDNA libraries of E. coli cells, the group of D. J. Cahill (Max Plank Institute of Molecular Genetics) developed highly dense protein chips, which are used for selection of monoclonal antibodies and their synthetic analogs (aptamers, AffibodyTM, etc.), for searching of novel partners in protein-protein interactions, and also in complex experiments on transcriptional and proteomic mapping [80]. Chips produced by WITA Proteomics may be used for analysis of toxic effects on cell cultures or laboratory animals during pharmacological screening. (These chips employ 2D-electrophoresis.) This company also created a virtual library of expression of protein-intermediates of toxic effects on proteomic 2D-maps [81].
Protein biochips represent relatively young but intensively developed branch of proteomic technology, which may “drive” this science towards practical needs of medical diagnostics and design of new medical drugs.
Interactive proteomics. Studies of protein-protein interactions and supramolecular complex formation represent one of the main directions of functional proteomics. The importance of this direction cannot be overestimated because most biochemical reactions in the cell are structure-related and enzymes, receptor, and biologically active compounds act as supramolecular assemblies [82-84]. A yeast two-hybrid system is widely used for the determination of partner couple of protein-protein interactions [84] (Fig. 4).
This system is one of the main modes for searching and study of protein-protein, protein-DNA, and protein-RNA interactions. This system is quite reliable in a wide range of experiments from analysis of certain complexes to creation of networks of protein interactions of whole organisms. The yeast two-hybrid system represents a continuation of the logic of studies on transcription activators. Site-specific activators often contain DNA-binding domain (BD) and transcription activation domain (AD). According to the original scheme of two-hybrid system two putative interacting proteins are constructed into hybrid (chimeric) proteins bound to BD and AD, respectively. Both hybrid proteins BD-X and AD-Y are co-expressed by the yeast strain carrying specific DNA-site for transcription activator. This site is located in 5´-flanking region of some (reporter) gene, whose expression can be easily detected in growing yeast cells.Fig. 4. Yeast two-hybrid system. X and Y are two interacting proteins; BD and AD are DNA-binding domain and transcription activator domain, respectively.
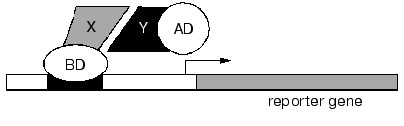
The interaction of X and Y proteins restores the structure of the transcription activator; this results in transcription of the reporter gene (Fig. 4) and to characteristic cell phenotype.
The yeast two-hybrid system is often used for determination of interaction of known protein partners and also for screening of genomic libraries (e.g., for search of potential partners which would interact with a certain protein).
The two-hybrid system may also be used for characterization of networks of protein-protein interactions. For this purpose a wide screening of each protein expressed by a eukaryotic cell is used. Such approach may reveal some unexpected links between certain cellular processes and genes with poorly characterized functions.
The original yeast two-hybrid system has some limitations because some (e.g., membrane) proteins cannot be reconstituted in the nucleus or they require additional factors for modification. Studies of interactions of membrane proteins employ alternative non-transcriptional two-hybrid (cytoplasmic) systems.
Such a two-hybrid system can use small ligand molecules for pharmacological studies (wide screening).
Recently, so-called biomolecular interactive analysis (BIA) has become rather popular among scientists studying protein-protein interactions. BIA evaluates protein-protein interaction in the real time mode on the surface or in the cuvette of optical biosensor without any modification of the interaction partners. During the last decade this technology has been significantly modified because it was coupled to mass-spectrometry for identification of proteins after the registration of molecular event [85, 86]. Several multichannel optical biosensors are known. They employ the principle of resonant mirror or plasmon resonance, which allows monitoring of highly sensitive interactions between proteins (fishing) and provides material for mass spectrometry in the online mode. The combination of modern optical biosensors with microfluid technology allows screening of interacting couples and assemblies of biological molecules in the automated mode [87]. This approach can be used to obtain kinetic characteristics of protein-protein interactions and to register not only potent but also weak intermolecular interactions of multicomponent systems in dependence on conditions of interaction medium (ion concentration, pH, etc.). This approach does not have limitations for analysis of membrane protein interactions, and it will probably displace the two-hybrid system in the future.
BIOINFORMATICS FOR PROTEOMICS
Databases for 2D-gel images. Due to increase in developments in the art of 2D-electrophoresis exchange of scientific information and useful accessibility became necessary. So, in 1996 integrated databases of 2D-electrophoretic maps and programs for the processing of images of these maps were developed using bioinformatic methods [88].
The integrated databases should meet the following criteria: each entry in the database should be available during search by key words; the database should be connected to other databases via active links; an entry in the database should contain the main index (which is the index of SWISS-PROT); in this database bi-directional links between the main index and other indices should exist; each entry in the database should be available in response to a “mouse click” on the protein spot of the image. Programs for image analysis developed for the work with the integrated database should provide direct access of individual entries into any integrated database.
Some existing databases are listed in Table 5. SWISS-2DPAGE is the most popular database. Besides images of 2D-gels with identified proteins on them and such experimental data as isoelectric point, molecular mass, this database contains information obtained during comparison of gels, microsequencing, immunoblotting, analysis of amino acid composition, and mass-spectrometry analysis of peptide fingerprints. This database also contains bibliographic references and various links on other biological servers.
Table 5. Databases of 2D-electrophoretic
maps

Two special catalogues of numerous databases of 2D-gel electrophoretic maps have been prepared. The first one, WORLD-2DPAGE (http://www.expasy.ch/ch2d/2d-index.html), contains references on known databases of 2D-electrophoresis images and equipment for their treatment. The second database, 2DWG Meta-Database (http://www-lecb.ncifcrf.gov/2dwgDB) [89], was organized as a large electronic table. Each row of this table provides data corresponding to a gel; it also includes a type of tissue, references on gel image and on database. Also there is a special search system for Internet-resources of maps of 2D-electrophoresis, BioHunt (http://www.expasy.ch/BioHunt).
Programs for comparison of 2D-proteomic maps. Under carefully standardized conditions the position of a protein on gel is sufficient for its identification. Matching of maps of 2D gels obtained in experiments with previously characterized maps with identified proteins from databases or comparison of control and experimental gels may allow the determination of proteins expressed under certain environmental conditions, stress treatments, or in some pathological processes.
Corresponding algorithms for gel matching can be subdivided into two groups: 1) algorithms based on characteristics of spot image on gel; 2) algorithms based on direct comparison of images by distribution of intensity.
The algorithms of the first group are used in such programs as PDQuest (http://www.proteomeworks.bio-rad.com/index.htm), Phoretix 2D (http://www.phoretix.com/products/2d products.htm), Melanie (http://www.expasy.ch/melanie); they are used in numerous proteomic studies [90]. The standard procedure of gel image matching used in Melanie and other programs of this group consists of detection of spots and their editing, landmarking of compared gels, alignment and matching. Matching of gels begins with determination of clusters of neighboring spots |for each spot. Clusters corresponding to input data by cluster couples are matched first. In case of absence of input data clusters with the most intensive primary spot (primary clusters) are first matched. After that clusters are compared by using homologous measure, which is based on the probability approach, where in the first approximation probability of accidental coincidence is calculated. During the next stage secondary clusters are compared. The last stage includes verification of results (and exclusion of wrong comparisons). After that for each three spots in each primary cluster transformation rules are calculated; these rules are subsequently used for comparison and determination of paired spots on the compared gels [91].
Algorithms based on direct comparison of images by distribution of intensity are used in such programs as Z3 (http://www.2dgels.com), MIR (http://www.doc.ic.ac.uk/~gzy). These programs employ on original approach for complementary pseudocolor staining of the comparable gels (e.g., with purple or green color). In this case superimposition of the compared gels the overlapping spots give gray or black color (depending on protein amount) [92, 93].
The algorithm used by Z3 is based on comparison of small regions of gel images containing from two to four protein spots with unique pattern followed by subsequent calculation of image transformation as a shift vector [92].
Program MIR uses an approach of multilevel comparison of gels. The main idea is that image distortions leading to insufficient alignment may be decomposed into constituents by their corresponding components at each level of resolution. At low resolution of gel only rough distortions can be visible and rough approximation for optimal geometric transformation of the image may be calculated. After the removal of these rough distortions more gentle distortions will remain at the next stage of resolution. Such procedure may be carried by recursive mode for final optimization of geometrical transformation of the image [93].
Some programs allow comparison of images of 2D-electrophoresis and identification of proteins in the Internet using Java-supplement. These include FLICKER (http://www.lecb.ncifcrf.gov/flicker) and CAROL (http://www.gelmatching.inf.fu-berlin.de). Such programs have certain advantages: low cost of the existing technology, which does not require special efforts of user, rather short time for analysis than in the case of other programs for 2D-gel matching. The use of such programs also provides almost unlimited access to the integrated databases. However, these programs are available only on servers where they are located. For security reasons most servers have limited access to resources of the server on which such supplement is available.
Programs and algorithms used for identifications of mass-spectra. The general principle for the protein identification by mass spectrometry consists of matching of experimentally obtained mass spectrum with database mass spectra of known proteins. In proteomic studies mass-spectrometry identification of proteins separated by 2D-electrophoresis and subjected to enzymatic proteolysis is used. The mass spectrometry pattern of products of proteolytic digestion of the analyzed protein (peptide fingerprint) is unique. Using search algorithm the resultant pattern may be compared with the theoretical mass-spectrum. Various search algorithms are characterized by certain properties and knowledge of these properties is important for final results.
The use of tandem mass spectrometry (MS/MS) significantly increases effectiveness of mass-spectrometry. The ionized peptides selected by mass in the first mass spectrometer are subjected to the subsequent fragmentation during collision with a gas (in the second mass spectrometer) and the resultant peptides are used for determination of amino acid sequence. Using database query by known peptide sequence the analyzed protein may be identified. Employing tandem mass spectrometry as useful research tool user should be confident that information (fragments of peptide sequences, complex protein mixtures, etc.) on components of the research objects is available in EST-databases.
As a rule, programs for protein identification by mass spectra use freely available databases containing amino acid sequences of proteins and nucleotide sequences of genes. Below we consider the most widely known databases:
- SWISS-PROT is a database of annotated protein sequences; it also contains additional information on function of the protein, its domain structure, posttranslational modification(s), etc.;
- TrEMBL is a supplement to SWISS-PROT, which contains all protein sequences, translated from nucleotide sequences of the EMBL database;
- PIR-International (Protein Identification Resource, National Biomedical Research Foundation, Washington, USA) is also an annotated database of protein sequences;
- NCBInr (National Center of Biotechnological Information) is a database containing sequences translated from DNA sequences of GenBank and also sequences from PDB, SWISS-PROT, and PIR databases;
- ESTdb (Expressed Sequence Tags database, NCBI, NIH).
These databases are constantly updated and are usually characterized by the standardized data format.
The existing algorithms and corresponding programs may be subdivided into three main groups:
- programs using proteolytic peptide fingerprint for protein identification (PeptIdent, MultiIdent, ProFound);
- programs additionally operating with MS/MS spectra (PepSea, MASCOT, MS-Fit, MOWSE) or with MS/MS only;
- programs operating with MS/MS only (SEQUEST, PepFrag, MS-Tag, Sherpa).
The simplest algorithms for protein search in database matches experimentally obtained and theoretical mass spectra and ranks database proteins according to number of matching peptides. Such algorithms often overestimate large proteins, for which a greater number of calculated peptides is possible (this increases probability of random matching).
More perfect algorithms use additional information such as isoelectric point of protein, its molecular mass, amino acid composition, etc. (PeptIdent, MultiIdent) [94].
The algorithm MOWSE [95] is more selective and sensitive than other algorithms calculating only number of matching peptides. Calculating estimates it takes into consideration both relative excess of peptides of the same molecular mass in the database and size of protein.
Program MASCOT [96] is based on the MOWSE algorithm; this program also evaluates a possibility of random matching of experimental and theoretical peptide masses. Using sequential queries MASCOT can optimize search parameters. Using this program it is possible to identify proteins in relatively simple mixtures.
Some algorithms may rank results by probability of correspondence to the query. For example, program ProFound [97] uses Bayesian statistics for ranking proteins in the database, which minimizes expected probability of wrong classification. This program uses a step-by-step approach for protein identification. Initially, database proteins are ranked according to their matching with experimental data using assumption that there is only one protein in the query. Then mass spectra of peptides from proteins that received the highest estimate are pooled by two, three, etc. Such pooled spectra are ranked again by similarity with query. Similar algorithm allows identification of a few proteins by their fingerprints.
Program SEQUEST is for identification of proteins on the basis of mass spectra of fragment ions; it calculates correlation coefficient between experimental mass spectra and those calculated by sequences from databases. Estimates for database proteins are formed on the basis of this correlation coefficient. Besides fragment ion spectra, this approach does not require any other information, but the search is time consuming. Program Sherpa [98] has insignificant differences from SEQUEST.
Since protein identification by tandem mass spectrometry is a rather lengthy procedure, this method is not widely used in proteomic studies as mass-spectrometry of peptide fragments. So, comparative analysis of effectiveness of algorithms identifying proteins by their fingerprint is of special interest.
Comparative analysis of algorithm effectiveness employed the theory of signal detection [99]. Within the framework of this theory, correct identification and random matching are characterized by various distribution. The most popular algorithms such as Bayesian (ProFound), MOWSE (modified), program MS-FIT, MOWSE (probability based) (program Mascot), and Number of Matches (program MS-Fit) were comparatively analyzed for four proteins (yeast 25 kD, mouse 50 kD, human 100 kD, and Caenorabditis elegans 200 kD). Over the whole range of protein masses Bayesian algorithm demonstrates the best results versus other algorithms. The best results for the algorithms MOWSE (modified) were within rather low range region of masses within 50 kD, whereas for the algorithms MOWSE they did not exceed 100 kD.
Methodology of proteomic studies is simultaneously developed in several directions and significantly influences our notions on the capabilities of biological sciences. Using databases on complete nucleotide sequences of genomes, methods of protein sequencing de novo by mass spectrometry, and highly effective methods of protein separation by means of 2D-electrophoresis and chromatography significantly increases abilities of certain biological experiment and gives a fast algorithm for decoding particular molecular mechanisms and new features of functioning of the living matter. One of the important modern problems is the development of the metabolon. The latter represents a new biochemical notion describing integrated assembly of reactions and interaction of macromolecules within the cell, tissue, organ, and organism. This will attract much interest during the coming decade. Protein inventory is one of the first stages in metabolon building. We do realize that the modern methods used in proteomic studies are not sufficient for exhaustive solution of this problem. However, modern results inspire certain optimism. It is also important that commercial organizations and governmental structures in many countries all over the world pay much attention to proteomics. The development of this technology may provide the “long pass break” in design of new highly effective drugs, introduction of novel approaches into diagnostics of various diseases, and discoveries of new principles of cell functioning. The annual investments into this field are estimated as a billion dollars. In contrast to other biotechnological branches of science, proteomics has been being developed in Russia during the last decade. Last year the Center Proteomic Studies was organized at Orekhovich Institute of Biomedical Chemistry, Russian Academy of Medical Sciences. This center has modern resources for proteomic service. The power of this Center allows up to 100 2D-electrophoresis weekly followed by protein identification by the methods of MALDI and tandem mass spectrometry. We believe that Russian developments in this filed will represent a valuable contribution into the world proteomic program.
Authors are grateful to members of the stuff of Center of Proteomic Studies at Orekhovich Institute of Biomedical Chemirtsy, Russian Academy of Medical Sciences, O. V. Tikhonova, S. A. Moshkovsky, P. G. Lokhov, and M. V. Serebryakova for their valuable help and constructive criticism during preparation and discussion of this review.
REFERENCES
1.Wasinger, V. C., Cordwell, S. J., Cerpa-Poljak, A.,
Yan, J. X., Gooley, A. A., Wilkins, M. R., Duncan, M. W., Harris, R.,
Williams, K. L., and Humphery-Smith, I. (1995) Electrophoresis,
16, 1090-1094.
2.Anderson, N. G., and Anderson, L. (1982) Clin.
Chem., 28, 739-748.
3.Humphery-Smith, I., Cordwell, S. J., and
Blackstock, W. P. (1997) Electrophoresis, 18,
1217-1242.
4.Wilkins, M. R., Williams, K. L., Appel, R. D., and
Hochstrasser, D. F. (1997) Proteome Research: New Frontiers in
Functional Genomics, Springer, Berlin.
5.Blackstock, W. P., and Weir, M. P. (1999)
TIBTECH, 17, 121-127.
6.Service, R. F. (2000) Science, 287,
2136-2138.
7.Champion, K. M., Nishihara, J. C., Joly, J. C., and
Arnott, D. (2001) Proteomics, 1, 1133-1148.
8.Petersohn, A., Brigulla, M., Haas, S., Hoheisel, J.
D., Volker, U., and Hecker, M. (2001) J. Bacteriol., 183,
1517-1531.
9.Buttner, K., Bernhardt, J., Scharf, C., Schmid, R.,
Mader, U., Eymann, C., Antelmann, H., Volker, A., Volker, U., and
Hecker, M. (2001) Electrophoresis, 22,2908-2935.
10.Tonella, L., Hoogland, C., Binz, P. A., Appel, R.
D., Hochstrasser, D. F., and Sanchez, J. C. (2001) Proteomics,
1, 409-423.
11.Anderson, L. N., Matheson, A. D., and Steiner, S.
(2000) Curr. Opin. Biotechnol., 11, 408-412.
12.Gygi, S. P., Rochon, Y., Franza, B. R., and
Aebersold, R. (1999) Mol. Cell. Biol., 19, 1720-1730.
13.Knezevic, V., Leethanakul, C., Bichsel, V. E.,
Worth, J. M., Prabhu, V. V., Gutkind, J. S., Liotta, L. A., Munson, P.
J., Petricoin III, E. F., and Krizman, D. B. (2001) Proteomics,
1,1271-1278.
14.Adam, B. L., Vlahou, A., Semmes, O. J., and
Wright, G. L., Jr. (2001) Proteomics, 1, 1264-1270.
15.Wulfkuhle, J. D., McLean, K. C., Paweletz, C. P.,
Sgroi, D. C., Trock, B. J., Steeg, P. S., and Petricoin III, E. F.
(2001) Proteomics, 1, 1205-1215.
16.Emmert-Buck, M. R., Bonner, R. F., Smith, P. D.,
Chuaqui, R. F., Zhuang, Z., Goldstein, S. R., Weiss, R. A., and Liotta,
L. A. (1996) Science,274,998-1001.
17.Jones, M. B., Krutzsch, H., Shu, H., Zhao, Y.,
Liotta, L. A., Kohn, E. C., and Petricoin III, E. F. (2002)
Proteomics, 2, 76-84.
18.Molloy, M. P. (2000) Analyt. Biochem.,
280, 1-10.
19.Herbert, B. R., Molloy, M. P., Gooley, A. A.,
Walsh, B. J., Bryson, W. G., and Williams, K. L. (1998)
Electrophoresis, 19, 845-851.
20.Ruegg, U. T., and Rudinger, J. (1977) Meth.
Enzymol., 48,111-117.
21.MacLaren, J. A. (1971) Text. Res. J.,
41, 713.
22.Rabilloud, T., Adessi, C., Giraudel, A., and
Lunardi, J. (1997) Electrophoresis, 18, 307-316.
23.Perdew, G. H., Schaup, H. W., and Selivonchick,
D. P. (1983) Analyt. Biochem., 135, 453-455.
24.Pasquali, C., Fialka, I., and Huber, L. A. (1997)
Electrophoresis, 18,2573-2581.
25.Santoni, V., Rabilloud, T., Doumas, P., Rouquie,
D., Mansion, M., Kieffer, S., Garin, J., and Rossignol, M. (1999)
Electrophoresis, 20, 705-711.
26.Molloy, M. P., Herbert, B. R., Slade, M. B.,
Rabilloud, T., Nouwens, A. S., Williams, K. L., and Gooley, A. A.
(2000) Eur. J. Biochem., 267,2871-2881.
27.Fujiki, Y., Hubbard, A. L., Fowler, S., and
Lazarow, P. B. (1982) J. Cell. Biol., 93, 97-102.
28.Kamo, M., Kawakami, T., Miyatake, N., and
Tsugita, A. (1995) Electrophoresis, 76, 423-430.
29.Garrels, J. I., Me Laughlin, C. S., Warner, J.
Ft., Futcher, B., Latter, G. I., Kobayashi, R., Schwender, B., Voipe,
T., Anderson, D. S., Mesquita-Fuentes, R., and Payne, W. E. (1997)
Electrophoresis, 18, 1347-1360.
30.Perrot, M., Sagliocco, F., Mini, T., Monribot,
C., Schneider, U., Shevchenko, A., Mann, M., Jeno, P., and Boucherie,
H. (1999) Electrophoresis, 20, 2280-2298.
31.Costa, P., Pionneau, C., Bauw, G., Dubos, C.,
Bahrmann, N., Kremer, A., Frigerio, J. M., and Plomion, C. (1999)
Electrophoresis, 20, 1098-1108.
32.Santoni, V., Rouquie, D., Doumas, P., Mansion,
M., Boutry, M., Degand, H., Dupree, P., Packman, L., Sherrier, J.,
Prime, T., Bauw, G., Posada, E., Rouze, P., Dehais, P., Sahnoun, I.,
Barlier, I., and Rossignol, M. (1998) Plant. J.,
16,633-641.
33.Link, A. J., Eng, J., Schieltz, D. M., Carmack,
E., Mize, G. J., Morris, D. R., Garvik, B. M., and Yates III, J. R.
(1999) Nat. Biotechnol., 17, 676-682.
34.Mirgorodskaya, O. A., Kozmin, Y. P., Titov, M.
I., Korner, R., Sonksen, C. P., and Roepstorff, P. (2000) Rapid
Commun. Mass Spectrom., 14, 1226-1232.
35.O'Farrell, P. H. (1975) J. Biol. Chem.,
250, 4007-4021.
36.Link, A. J. (1999) 2-D Proteome Analysis
Protocols, Humana Press, Totova, New Jersey.
37.Anderson, N. G., and Anderson, N. L. (1978)
Analyt. Biochem., 85, 331-354.
38.Dunn, M. J. (1993) Gel Electrophoresis of
Proteins, BIOS Scientific Publishers Ltd. Alden Press, Oxford.
39.Görg, A., Postel, W., Westermeier, R.,
Gianazza, E., and Righetti, P. G. (1980) J. Biochem. Biophys.
Meth., 3, 273-284.
40.Görg, A., Postel, W., and Günther, S.
(1988) Electrophoresis, 9, 531-546.
41.Görg, A., Postel, W., Friedrich, C., Kuick,
R., Strahler, J. R., and Hanash, S. M. (1991) Electrophoresis,
12, 653-658.
42.Laemmli, U. K. (1970) Nature, 227,
680-685.
43.Bjellqvist, B., Ek, K., Righetti, P. G.,
Gianazza, E., Görg, A., Westermeier, R., and Postel, W. (1982)
J. Biochem. Biophys. Meth., 6, 317-339.
44.Görg, A. (1993) Biochem. Soc. Trans.,
21, 130-132.
45.Matsui, N. M., Smith-Beckerman, D. M., Fichmann,
J., and Epstein, L. B. (1999) in 2-D Proteome Analysis
Protocols, Humana Press, Totova, New Jersey, p. 211.
46.Futcher, B., Latter, G. I., Monardo, P.,
McLaughlin, C. S., and Garrels, J. I. (1999) Mol. Cell. Biol.,
19, 7357-7368.
47.Gygi, S. P., Corthals, G. L., Zhang, Y., Rochon,
Y., and Aebersold, R. (2000) Proc. Natl. Acad. Sci. USA,
15, 9390-9395.
48.Herbert, B. R., and Righetti, P. G. (2000)
Electrophoresis, 21, 3639-3648.
49.Herbert, B. R., Harry, J. L., Packer, N. H.,
Gooley, A. A., Pedersen, S. K., and Williams, K. L. (2001) Trends
Biotechnol., 19 (Suppl.), s3-s9.
50.Nesterenko, M. V., Tilley, M., and Upton, S. J.
(1994) J. Biochem. Biophys. Meth., 28,239-242.
51.Shevchenko, A., Wilm, M., Vorm, O., and Mann, M.
(1996) Analyt. Chem., 68, 850-858.
52.Neuhoff, V., Arold, N., Taube, D., and Ehrhardt,
W. (1988) Electrophoresis, 9, 255-262.
53.Yan, J. X., Harry, R. A., Spibey, C., and Dunn,
M. J. (2000) Electrophoresis, 21, 3657-3665.
54.Bilban, M., Buehler, L. K., Head, S., Desoye, G.,
and Quaranta, V. (2002) Curr. Issues Mol. Biol., 4,
57-64.
55.Westermeier, R. (2001) in Proc. Proteomic
Forum 2001, Technische Universitat Muenchen, Germany, p. 53.
56.Yates III, J. R., Link, A. J., and Schieltz, D.
(2000) Meth. Mol. Biol., 146, 17-26.
57.Washburn, M. P., Wolters, D., and Yates III, J.
R. (2001) Nat. Biotechnol., 19, 242-247.
58.Gevaert, K., and Vandekerckhove, J. (2000)
Electrophoresis, 21, 1145-1154.
59.Wolters, D. A., Washburn, M. P., and Yates III,
J. R. (2001) Analyt. Chem., 73, 5683-5690.
60.Gygi, S. P., Rist, B., Gerber, S. A., Turecek,
F., Gelb, M. H., and Aebersold, R. (1999) Nat. Biotechnol.,
17, 994-999.
61.Moseley, M. A. (2001) Trends Biotechnol.,
19 (Suppl.), s10-s16.
62.Aebersold, R., and Goodlett, D. R. (2001)
Chem. Rev., 101, 269-295.
63.Nordhoff, E., Egelhofer, V., Giavalisco, P.,
Eickhoff, H., Horn, M., Przewieslik, T., Theiss, D., Schneider, U.,
Lehrach, H., and Gobom, J. (2001) Electrophoresis, 22,
2844-2855.
64.Roepstorff, P. (2000) EXS,88,
81-97.
65.Gygi, S. P., and Aebersold, R. (2000) in Using
Mass-Spectrometry for Quantitative Proteomics. Proteomics: A Trends
Guide, pp. 31-36.
66.Andersen, J. S., and Mann, M. (2000) FEBS
Lett., 480, 25-31.
67.Kussmann, M., and Roepstorff, P. (2000) Meth.
Mol. Biol., 146, 405-424.
68.Chaurand, P., Luetzenkirchen, F., and Spengler,
B. (1999) J. Am. Soc. Mass Spectrom., 10, 91-103.
69.Spengler, B. (1997) J. Mass Spectrom.,
32, 1019-1036.
70.Jonscher, K. R., and Yates III, J. R. (1997)
Analyt. Biochem., 244, 1-15.
71.Qin, J., and Chait, B. T. (1997) Analyt.
Chem., 69, 4002-4009.
72.Wilm, M., and Mann, M. (1996) Analyt.
Chem., 68, 1-8.
73.Papayannopoulos, I. A. (1995) Mass Spectrom.
Rev., 14, 49-73.
74.Hillenkamp, F., Karas, M., Beavis, R. C., and
Chait, B. T. (1991) Analyt. Chem., 63, 1193A-1203A.
75.Fenn, J. B., Mann, M., Meng, C. K., Wong, S. F.,
and Whitehouse, C. M. (1990) Mass Spectrom. Rev.,
9,37-70.
76.Twerenbold, D., Gerber, D., Gritti, D., Gonin,
I., Netuschill, A., Rossel, F., Schenker, D., and Vuilleumier, J.-L.
(2001) Proteomics, 1, 66-69.
77.Wright, G. L., Cazares, L. H., Leung, S. M.,
Nasim, S., Adam, B., Yip, T., Schellhammer, P. F., Gong, L., and
Vlahou, A. (1999) Prost. Cancer Prost. Dis., 2,
264-276.
78.Vlahou, A., Schellhammer, P. F., Mendrinos, S.,
Patel, K., Kondylis, F. I., Gong, L., Nasim, S., and Wright, G. L., Jr.
(2001) Am. J. Pathol., 158, 1491-1502.
79.He, M., and Taussig, M. J. (2001) Nucleic
Acids Res., 29, e73.
80.Cahill, D. J. (2001) J. Immunol. Meth.,
250, 81-91.
81.Bolon, P. (2002) in Protein and Peptide
Arrays: Pitfalls, Prospects, and Promise, 26-27
March, Munich, Germany.
82.Rain, J.-C., Selig, L., De Reuse, H., Battaglia,
V., Reverdy, C., Simon, S., Lenzen, G., Petel, F., Wojcik, J.,
Schaechter, V., Chemama, Y., Labigne, A., and Legrain, P. (2001)
Nature, 409, 211-215.
83.Uetz, P., Giot, L., Cagney, G., Mansfield, T. A.,
Judson, R. S., Knight, J. R., Lockshon, D., Narayan, V., Srinivasan,
M., Pochart, P., Qureshi-Emili, A., Li, Y., Godwin, B., Conover, D.,
Kalbfleisch, T., Vijayadamodar, G., Yang, M., Johnston, M., Fields, S.,
and Rothberg, J. M. (2000) Nature, 403, 623-627.
84.Ito, T., Chiba, T., Ozawa, R., Yoshida, M.,
Hattori, M., and Sakaki, Y. (2001) Proc. Natl. Acad. Sci.
USA,98, 4569-4574.
85.Natsume, T., Nakayama, H., and Isobe, T. (2001)
Trends Biotechnol., 19 (Suppl.), s28-s33.
86.Malmqvist, M., and Karlsson, R. (1997) Curr.
Opin. Chem. Biol.,1, 378-383.
87.Szabo, A., Stolz, L., and Granzow, R. (1995)
Curr. Opin. Struct. Biol., 5, 699-705.
88.Appel, R. D., Bairoch, A., Sanchez, J. C.,
Vargas, J. R., Golaz, O., Pasquali, C., and Hochstrasser, D. F. (1996)
Electrophoresis, 17, 540-546.
89.Lemkin, P. F. (1997) Electrophoresis,
18, 2759-2773.
90.Pleissner, K. P., Oswald, H., and Wegner, S.
(2001) in Proteomics. From Protein Sequence to Function
(Pennington, R., and Dunn, M. J., eds.) BIOS Scientific Publishers,
Oxford, pp. 131-149.
91.Appel, R. D., Vargas, J. R., Palagi, P. M.,
Walther, D., and Hochstrasser, D. F. (1997) Electrophoresis,
18, 2735-2748.
92.Smilansky, Z. (2001) Electrophoresis,
22, 1616-1626.
93.Veeser, S., Dunn, M. J., and Yang G.-Z. (2001)
Proteomics, 1, 856-870.
94.Wilkins, M. R., Gasteiger, E., Wheeler, C.,
Lindskog, I., Sanchez, J.-C., Bairoch, A., Appel, R. D., Dunn, M. D.,
and Hochstrasser, D. F. (1998) Electrophoresis, 19,
3199-3206.
95.Pappin, D. J. C., Hojrup, P., and Bleasby, A. J.
(1993) Curr. Biol., 3, 327-332.
96.Perkins, D. N., Pappin, D. J. C., Creasy, D. M.,
and Cottrell, J. S. (1999) Electrophoresis, 20,
3551-3567.
97.Zhang, W., and Chait, B. T. (2000) Analyt.
Chem., 72, 2482-2489.
98.Taylor, J. A., Walsh, K. A., and Johnson, R. S.
(1996) Rapid Commun. Mass Spectrom., 10, 679-687.
99.Green, D. M., and Swets, J. A. (1966) in
Signal Detection Theory and Psychophysics, Wiley, Reprinted 1974
by Krieger, Huntington, New York.