Endotoxin-Lipoprotein Complex Formation as a Factor in Atherogenesis: Associations with Hyperlipidemia and with Lecithin:Cholesterol Acyltransferase Activity
Y. Sh. Schwartz1,2* and M. I. Dushkin1,2
1Institute of Internal Medicine, Siberian Division of the Russian Academy of Medical Sciences, ul. Vladimirovskii Spusk 2a, Novosibirsk, 630003 Russia; fax: (3832) 29-32532Institute of Clinical Immunology, Siberian Division of the Russian Academy of Medical Sciences, ul. Yadrintsevskaya 14, Novosibirsk, 630090 Russia; fax: (3832) 22-7028; E-mail: YShSchwartz@mail.ru
* To whom correspondence should be addressed.
Received December 10, 2001; Revision received March 27, 2002
A potential role of endotoxin-lipoprotein (bacterial lipopolysaccharide-lipoprotein, LPS-LP) complex formation as a pathogenic factor for atherosclerosis has not been studied yet. The aim of this study was to test the hypothesis that in endotoxinemia in humans hyperlipidemia associated with atherosclerosis development can favor an excessive LPS-LP complex formation, and endotoxin presented in blood can inhibit lecithin:cholesterol acyltransferase (LCAT), one of the key enzymes of reverse cholesterol transport. Endotoxin-binding capacity of lipoproteins (LP) in patients with normolipidemia and hyperlipidemia types IIa and IV was estimated from label incorporation into different LP fractions isolated by means of sequential ultracentrifugation following serum preincubation with Salmonella minnesota R595 125I-labeled LPS. The effect of varied concentrations of S. minnesota R595 LPS on LCAT activity was evaluated from the overall esterifying activity of serum using [1,2-3H2]cholesterol-labeled substrate. The elevation of low density LP (LDL) and very low density LP (VLDL) contents in blood serum in hyperlipidemia types IIa and IV, respectively, resulted in significant elevation of LPS binding to these fractions. LPS added to the blood serum leads to the dose-dependent decrease in LCAT activity. The revealed phenomena of elevated LPS binding to atherogenic LP fractions in hypercholesterolemia and endotoxin-induced LCAT inhibition suggest the pathogenic role of LPS-LP complexes in atherogenesis.
KEY WORDS: atherosclerosis, endotoxin, lipoproteins, endotoxin-binding capacity, endotoxin-lipoprotein complex, lecithin:cholesterol acyltransferase
Abbreviations: LCAT) lecithin:cholesterol acyltransferase; LP) lipoprotein; LPS) lipopolysaccharide; LBP) LPS-binding protein; LDL) low density LP; VLDL) very low density LP; HDL) high density LP; CM) chylomicrons; CS) cholesterol; CETP) cholesterol ester transfer protein; TG) triglyceride; HLP) hyperlipidemia; PHMB) p-OH-methylbenzimidate.
The hypothesis that bacterial lipopolysaccharides (endotoxins) can
interact with blood serum lipoproteins (LP) arose in the mid 60s [1]. Since then numerous studies have been performed to
test and identify the participation of LP in lipopolysaccharide (LPS)
binding and inactivation. The great majority of investigators found
that normally high density lipoproteins (HDL) are the main LPS-binding
lipoproteins in blood sera of experimental animals. In particular,
Freudenberg et al. [2] used immunoelectrophoresis
to demonstrate that after intravenous LPS injection the blood serum
proteins with changed electrophoretic mobility were identified as HDLs,
and changes in their mobility resulted from complex formation between
them and LPS. Ulevitch and Johnston [3]
demonstrated by ultracentrifugation that the LPS buoyant density in
blood serum changes due to the presence of HDL, but neither low density
LP nor very low density LP (LDL and VLDL, respectively) caused this
effect. Usynin et al. [4] demonstrated an ability
of rat HDL3 (but not other LP fractions) to prevent
LPS-induced biochemiluminescence in macrophage culture. LPS-HDL
complexes were found in blood sera of various animals including humans.
The binding of LPS to HDL was shown to retard LPS clearance, inhibit
LPS binding to cells [2, 5],
and prevent development of LPS-induced lethal effects [6].
In spite of the well-described ability of HDL to bind and neutralize LPS in plasma, some studies led to contradictory results. According to the studies of Freudenberg and Galanos [7], LPS clearance rate in blood does not depend on its binding to HDL. Flegel et al. [8] found in experiments on macrophage cultures that LDLs (but not other LP) block LPS-induced cell activation. Van Lenten et al. [9] concluded that all main LP classes bind LPS in direct proportion to cholesterol contents in these particles, and HDL is the main lipoprotein LPS acceptor only in animals (rabbit or rat) in which HDL serves also as the main cholesterol carrier. Indeed, LPS was found preferably in LDL and VLDL fractions in rabbits on cholesterol-enriched diet as well as in rabbits with hereditary hyperlipidemia (Watanabe rabbits).
The role of triglyceride-rich apoE-containing LP in LP binding has also been demonstrated. Human chylomicrons (CM), VLDL, and their remnants, when preincubated with endotoxin, decrease significantly the lethal effect of LPS in mice sensitized to endotoxin [10]. Rensen et al. [11, 12] demonstrated on rats that human recombinant apoE binds effectively an endotoxin and possesses a prominent protective effect against lethal doses of LPS.
Thus, in humans not only HDL, but possibly other LP classes can bind endotoxin. Because LPS binding to lipoproteins leads to neutralization of the main endotoxic effects of LPS, LPS-LP complex formation is considered in the literature as one of the important protective mechanisms in endotoxinemia, gram-negative infections, and sepsis. However, the role of endotoxin-lipoprotein complex formation in atherogenesis has not yet been studied. We demonstrated earlier, using an experimental model of atherosclerosis, an accelerated formation of atherosclerotic injuries of aorta under chronic endotoxinemia conditions [13]. In accordance with our hypothesis [14], LPS-LP complex formation is not only a compensatory mechanism directed toward the inactivation of LPS in blood serum, but also may be the main pathogenetic factor of atherosclerosis. In particular, we suppose that hyperlipidemia associated with atherosclerosis development might serve as a factor favoring the formation of LP-LPS complexes in endotoxinemia. LPS binding to LDL is therewith to retard the clearance of these LP and to favor their modification, elevated arrival into the arterial wall, and pro-inflammatory effects of endotoxin in the area of LP-LPS complex accumulation in vessels. LPS binding to HDL also appears to cause various proatherogenic effects, in particular, may inhibit apoA-I-dependent lecithin:cholesterol acyltransferase (LCAT; EC 2.3.1.43) activity thus hindering the normal process of reverse cholesterol transport. In connection with this, in the study presented here we estimated the endotoxin-binding capacity of different fractions of human blood serum lipoproteins, dependence of LPS-binding capacity of these fractions on hyperlipidemia (HLP) type, and effect of LPS on LCAT activity.
MATERIALS AND METHODS
Blood sera. Blood sera were isolated from 18 volunteers with normolipidemia (total cholesterol (CS) <5.2, alpha-CS >0.9, triglycerides (TG) <2.3 mM), 17 patients with HLP IIa type, and 16 patients with HLP IV type (HLP classification by Frederickson). CS and TG were determined by enzymatic methods using standard commercial kits (Biocon, Germany). Criteria for the patient selection by HLP type were: for IIa, high levels of total CS (>7.5 mM) and beta-CS (>5.0 mM) with normal TG level; and for IV type, normal CS, severe triglyceridemia (>5.0 mM), and absence of CM. Sera pooled from 5-6 persons were used in each of 3-4 independent experiments for the estimation of both LPS binding capacity of LP and LCAT activity.
Radioactive iodination of LPS. To estimate LPS to LP binding, highly purified LPS from Salmonella minnesota R595 (kindly provided by Drs. M. Freudenberg and C. Galanos, Max-Planck Institute for Immunobiology, Freiburg, Germany) was labeled by radioactive iodination according to the method [15] with slight modifications. For this purpose, we synthesized p-OH-methylbenzimidate (PHMB) [16], and linked it to LPS by the incubation of 1 mg LPS with 1 ml 50 mM PHMB in 0.05 M borate buffer, pH 8.0, for 20 h at 37°C, then unbounded PHMB was removed by dialysis against eight liters of saline, pH 7.4, for 24 h at 4°C. The resulting PHMB-derivative of LPS, which is sensitive to radioiodination, was labeled with Na125I (Amersham-Pharmacia Biotech UK Ltd., UK; with no carrier, specific activity 100 mCi/ml) using chloramine T. To do this, Na125I (1.2 mCi), 50 µl 10-4 M KI, and 10 µl chloramine T (1 mg/ml) were added to 1 mg PHMB-LPS conjugate in 1 ml saline, the mixture was incubated for 30 min at room temperature, and the reaction was stopped with 20 µl sodium metabisulfite (1 mg/ml). Unbound Na125I was removed by dialysis against five liters of saline for three days at 4°C using six changes of dialyzate, until the total radioactivity within it became less than 0.1% of total radioactivity in the dialysis sack, which achieved 1.1*108 cpm/ml. The resulting specific activity of 125I-labeled PHMB-LPS was about 0.1 µCi/µg.
Determination of LPS-binding capacity of LP fractions. To estimate the effect of hyperlipidemia type on the endotoxin-binding capacity of different LP fractions, labeled LPS was incubated with blood sera of patients (25 µg/ml) for 30 min at 37°C. After the 125I-LPS-LP complexes were formed, the fractions of VLDL, LDL, HDL2, and HDL3 (buoyant density d = 1.006, 1.019-1.063, 1.08-1.125, and 1.125-1.215 g/ml, respectively) were isolated by sequential ultracentrifugation in KBr solutions according to the method of Lindgren [17]. Centrifugation was performed on a Beckman L8-M ultracentrifuge (USA) with Ti-80 fixed-angle rotor at 105,000g and 8°C for 20 h at a time. Radioactivity was determined in each fraction, then the fractions were dialyzed, and protein was determined by the Lowry method [18].
Determination of the incorporation of labeled cholesterol into cholesterol esters of blood serum. Effects of LPS on LCAT activity were estimated by esterifying activity of pooled sera from persons with normolipidemia, according to the method of Glomset [19] with minor modifications. For the substrate preparation, aliquots containing 5 MBq benzene solution of [1,2-3H2]cholesterol (Isotope, Russia) were evaporated under nitrogen flow and dissolved in 20 µl acetone, added microdropwise into 5 ml serum, and the label-containing serum was frozen. To determine LCAT activity, 0.2-ml aliquot of the serum tested was preincubated with S. minnesota Re LPS (0.1-100 µg/ml) for 30 min at 37°C, then 50 µl substrate was added, and the samples were incubated for 1 h at 37°C. Samples without LPS or incubation were taken as controls. Reaction was terminated with 20 volumes (5 ml) of the extracting mixture of chloroform and methanol (2 : 1 v/v) containing butyryl hydroxytoluene (10 µg/ml). After extraction the samples were mixed with five volumes of water, centrifuged for 10 min at 3,000 rpm; the lower phase containing lipids was re-sampled, evaporated in nitrogen at 60°C, dissolved in 20 µl chloroform, and analyzed by TLC on Silufol silica gel plates in the solvent system hexane-diethyl ether-acetic acid (90 : 10 : 1 v/v). The cut bands corresponding to cholesterol esters were transferred into scintillation liquid for the radioactivity measurements.
Statistical data processing was performed using common methods of variation statistics including calculations of the mean and standard deviation. The significance of differences between the means was estimated using Student's t-test.
RESULTS AND DISCUSSION
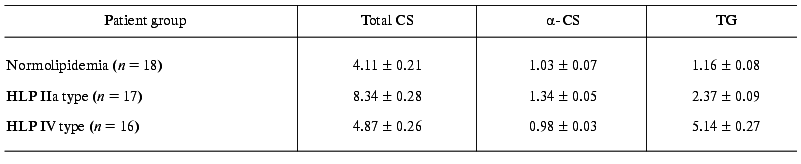
The mean level of lipids in blood sera used in experiments on dependence of endotoxin-binding capacity of different LP fractions on hyperlipidemia type is presented in the Table 1. As demonstrated, CS, alpha-CS, and TG contents in blood sera are in accordance with the criteria of HLP IIa and IV types.
Table 1. Total cholesterol (CS),
alpha-cholesterol (alpha-CS), and triglyceride (TG)
contents in blood sera of patients with normo- and hyperlipidemia (HLP)
(mM, M ± m)
The estimation of endotoxin-binding capacities of different LP fractions in blood sera of patients with normo- and hyperlipidemia shows that in any of the lipidemia types studied, LPs bind ~55-60% LPS (Table 2). In other words, both in norm and in hyperlipidemia the potential endotoxin-binding capacity of blood LP is very high and is higher than the overall capacity of all other LPS-binding components of serum, such as LPS-binding protein (LBP), bactericidal permeability increasing factor (BPI), soluble LPS receptor CD14 (sCD14), and others. In all lipidemia types under study, the highest (among LP fractions) LPS-binding level was observed in the HDL3 fraction, so in endotoxin-LP complexes formed the portion of LPS-HDL3 was 33-44% (Table 3). However, VLDL, LDL, and HDL2 also made significant contributions to LPS binding. Thus, total apoB-containing LP bound about the same amounts of LPS, as HDL3 in normolipidemia, and in hyperlipidemia the capacity of apoB-containing LPs was significantly higher than that of HDL3.
Table 2. Radioactivities of LP fractions
isolated after the incubation of blood serum with
125I-labeled LPS in patients with normo- and hyperlipidemia
(HLP) (A, cpm/ml serum (×106); B, cpm/mg protein
(×106))
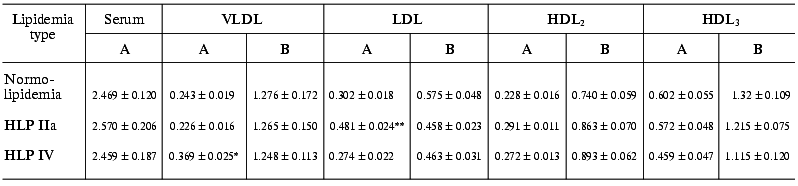
Note: The differences are significant with respect to the corresponding
controls in the normolipidemia group; * p < 0.01; ** p
< 0.001.
Table 3. Specific contributions of different
blood serum LP fractions to the 125I-labeled LPS binding in
patients with normo- and hyperlipidemia (A, % total radioactivity of
LPS in serum; B, % overall radioactivity of LPS in LP)
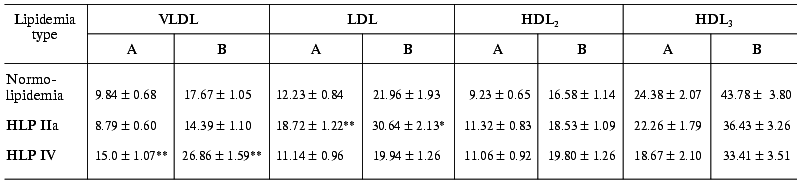
Note: The differences are significant with respect to the corresponding
controls in the normolipidemia group; * p < 0.05; ** p
< 0.001.
As we might expect, the portion of each fraction in LPS binding depended on the lipidemia type. In particular, in HLP IIa type characterized by hypercholesterolemia due to the extra-level of LDL, a significantly elevated level of LPS associated with the LDL fraction is observed: as compared to normolipidemia, the LPS portion in LDL 1.53-fold increases in relation to the total serum 125I-LPS level and 1.4-fold increases in relation to the 125I-LPS level in LPS-LP complexes (Table 3). In the similar way, in HLP IV type characterized by elevated triglyceride-rich LP level, VLDL-bound LPS portion 1.7-fold increases in relation to the total serum 125I-LPS level and 1.5-fold increases in relation to the level of 125I-LPS bound in LPS-LP complexes.
Thus, our data do not agree with many authors' opinion [4, 5, 20] about preferable LPS-binding activity of some individual LP class, because all LP fractions tested bound endotoxin effectively; an increase in portion of some fraction leads to corresponding increase in its capacity to bind LPS. Since a specific binding of endotoxin by LP fractions does not depend on lipidemia type (Table 4), it is obvious that the elevated ability of LDL and VLDL to bind LPS in corresponding HLP resulted from increased concentration of lipoprotein particles rather than from their increased affinity. In regards to atherogenesis, it is important that under hypercholesterolemia conditions the atherogenic LPs, such as LDL, can bind excessive amounts of endotoxins entering the blood. Hypercholesterolemia is known to be the main risk factor of atherosclerosis, and the increase in LDLs in blood leads to their elevated uptake and accumulation in arterial wall, to the stimulation of pro-inflammatory cytokine production in vessels, and a formation of atherosclerotic plaques and atheromas [21]. It is possible that the incorporation of LPS into LDL accelerates significantly this process [14, 22]. Unlike hypercholesterolemia, hypertriglyceridemia caused by elevated contents of blood VLDL level is not believed to be a direct factor of atherogenesis. However, numerous recent publications indicate a proatherogenic role of high VLDL level, which is particularly connected with enforced CETP-dependent substitution of cholesterol by TG in HDL, with a possibility of subfraction of small cholesterol-rich VLDL (VLDL2) to penetrate and detain in the subendothelial space, with a possibility of the direct uptake of large VLDL (VLDL1) by macrophages; and with a formation of highly atherogenic small dense LDLs from VLDL1 [23-26]. One may hypothesize that the proatherogenic effect of at least some of those mechanisms is enhanced in the presence of LPS in the VLDL composition [14].
Table 4. Specific LPS contents in different
LP fractions from blood sera of patients with normo- and hyperlipidemia
(µg/mg protein)
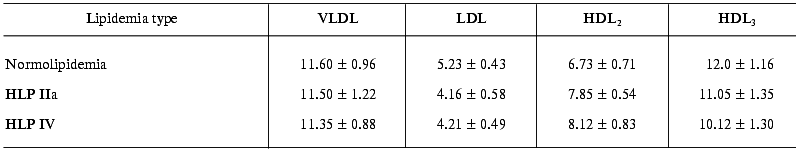
The data on the contribution of different LP classes to LPS-LP complex formation attract our attention because in spite of more than twofold decreased specific contents of LPS in LDL compared to VLDL (Table 4), the overall contribution of LDL to LPS binding was higher than that of VLDL (Table 3). The binding of LPS to LP obviously depends on lipoprotein particle amount at high endotoxin concentrations in blood. This dependence finds a logical explanation when taking into account that the amount of LDL particles in blood serum is considerably higher than that of VLDL.
As follows from our data, a specific LPS concentration in LPS-LP complexes varies from 4 to 12 µg per mg protein in different fractions (Table 4). This concentration is about one order lower than those reported by Victorov et al. [27, 28], who studied the mechanisms of complex formation between LDL and Salmonella typhimurium LPS. Since the presence of O-specific oligosaccharide chain in LPS molecule influences substantially the estimated LPS concentration, these discrepancies can be explained, because Re- and S-forms of LPS, respectively, were used in ours and the above mentioned experiments. The lower LPS concentrations and shorter incubation terms we used for the LPS binding to LP could also result in these differences.
It is unclear at present, what structural components of LDL and VLDL are responsible for their affinity to LPS. It was reported earlier that LPS-binding capability of LPs is determined by cholesterol [9] or other lipids [10] in their composition. An important role of apoproteins in the LPS-LP complex formation have been reported in ensuing years.
In particular, Berger et al. [29] reported that endotoxin neutralizing capability of blood serum depended directly on blood serum apoB concentration. The data of Rensen et al. [11, 12] indicate high endotoxin-binding capability of apoE. As for HDL, the ability of apoA-I to bind LPS was unequivocally demonstrated [4, 30, 31]. To take into account that Re-forms of LPS were used in the presented study, the certain endotoxin-binding capability found in LPs was apparently due to their interaction with lipid A. A presence of negatively charged phosphate groups in the lipid A molecule is logically suggested to allow it to interact with positively charged amino groups of lysine and arginine residues of apoprotein, and acyl residues of lipid A give it the possibility to form hydrophobic bonds with non-polar groups of amino acid residues of apoprotein. Quite recently, strong evidence was obtained for the incorporation of LPS into LP being mediated by LPS-binding protein (LBP) interacting effectively with apoB and circulating in blood in association with LDL and VLDL [20]. An ability of LBP to transfer catalytically LPS to apoA-I in reconstituted HDL was also demonstrated [31].
In endotoxinemia the blood lipoprotein spectrum is known to undergo a drastic alteration taking the appearance of an atherogenic spectrum. The HDL level diminishes significantly or remains unchanged, whereas the level of apoB-containing LPs many-fold increases [32-34]. For instance, Auerbach and Parks [34] reported that subcutaneous LPS injection into primates (300 µg per kg) induces after 48 h ~1.5-fold decrease in cholesterol level in HDL, decrease in apoA-I and apoA-II levels in HDL3a subfraction and a sharp increase in VLDL, as well as LDL levels accompanied by the elevation of plasma TG level up to 700%. As a result, a relative contribution of VLDL and LDL to the LPS-LP complex formation can obviously exceed significantly that for LPS-HDL. In the same time, the LPS-HDL complex formation can apparently lead to a proatherogenic result due to the negative effect on reverse cholesterol transport.
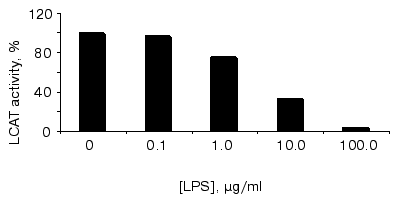
The anti-atherogenic role of HDLs is suggested to result preferably from two mechanisms: 1) their involvement in reverse cholesterol transport, namely in apoA-I-dependent acception of free cholesterol from peripheral tissues, its esterification on the HDL surface by LCAT, and subsequent transfer of some portion of cholesterol esters to apoB-containing LPs with a participation of cholesterol ester transfer protein (CETP); and 2) LCAT-dependent antioxidant effects of HDL particles [35]. LCAT inhibition affects both these anti-atherogenic mechanisms and can favor the development of atherosclerosis, whereas enhancement of LCAT activity prevents the development of atherosclerosis [36]. The effect of LPS on LCAT activity is shown in the figure. As one can see, endotoxin added to the blood serum exerts an inhibitory effect on LCAT activity in dose-dependent mode. In accordance to the data mentioned above [34], 48 h after LPS injection a certain decrease both in LCAT activity (by 55%) and LPL was observed in blood sera of primates with relative enrichment of all LP classes with phospholipids and TGs and significant decrease in CS ester contents. As it took place, HDL particles became discoid in shape and were enriched with apoE; and extremely high amounts of relatively nascent particles appeared in other LP classes. In other words, in endotoxinemia the blood LP composition becomes very much like that observed in blood sera of patients with hereditary LCAT deficiency. Our data suggest that LPS-dependent LCAT activity decrease can occur in vitro and, hence, is not connected obviously with this enzyme production in liver. Although the mechanisms of LCAT activity inhibition by LPS are unknown, the process of LPS-HDL complex formation appears to be one of them.
Earlier we provided some grounding for the hypothesis that the process of LPS detoxification in the course of interaction between endotoxin and blood lipoproteins leads finally to the initiation and progression of atheromatous injuries and is the main cause of atherosclerosis [14]. The phenomena we found in this study, namely elevated binding of LPS to atherogenic LP fractions in hypercholesterolemia and LPS-induced inhibition of LCAT, suggest this hypothesis.Effect of S. minnesota R595 LPS on the LCAT activity. To bind LPS to lipoproteins, varied concentrations of S. minnesota R595 LPS were incubated with blood serum for 30 min at 37°C, and then LCAT activity was determined as described in “Materials and Methods” section. Representative data of one of three independent experiments are shown. Esterifying activity in the absence of LPS was taken as 100%
This study was supported by the Russian Foundation for Basic Research (grant No. 00-04-49349).
REFERENCES
1.Skarnes, R. C. (1966) Ann. N. Y. Acad. Sci.,
133, 644-662.
2.Freudenberg, M. A., Bog-Hansen, T. C., Back, U.,
and Galanos, C. (1980) Infect. Immun., 28, 373-380.
3.Ulevitch, R. J., and Johnston, A. R. (1979) J.
Clin. Invest., 64, 1516-1524.
4.Usynin, I. F., Tsyrendorzhiev, D. D., Kharkovskii,
A. V., Polyakov, L. M., and Panin, L. E. (1996) Biochemistry
(Moscow), 61, 193-196.
5.Freudenberg, M. A., and Galanos, C. (1988)
Infect. Immun., 56, 1352-1357.
6.Levine, D. M., Parker, T. S., Donnely, T. M.,
Walsh, A., and Rubin, A. L. (1993) Proc. Natl. Acad. Sci. USA,
90, 12040-12044.
7.Freudenberg, M. A., and Galanos, C. (1992) in
Bacterial Endotoxic Lipopolysaccharides, Vol. 2,
Immunopharmacology and Pathophysiology (Ryan, J. L., and
Morrison, D. C., eds.) CRC Press, pp. 275-294.
8.Flegel, W. A., Baumstark, M. W., Weinstock, C.,
Berg, A., and Northoff, H. (1993) Infect. Immun., 61,
5140-5146.
9.Van Lenten, B. J., Fogelman, A. M., Haberland, M.
E., and Edwards, P. A. (1986) Proc. Natl. Acad. Sci. USA,
83, 2704-2708.
10.Harris, H. W., Grunfeld, C., Feingold, K. R., and
Rapp, J. H. (1990) J. Clin. Invest., 86, 696-702.
11.Rensen, P. C. N., van Oosten, M., van de Bilt,
E., van Eck, M., Kuiper, J., and van Berkel, Th. J. C. (1997)
Atherosclerosis, 134, 379.
12.Rensen, P. C. N., van Oosten, M., van Eck, M.,
van Amersfoort, E. S., van Dam, A.-M., Breve, J. J. P., Kuiper, J., and
van Berkel, Th. J. C. (1998) in Abst. V Ann. Scand.
Atherosclerosis Conf., Scandinavian Society for
Atherosclerosis Research, Humlebak, p. 42.
13.Schwartz, Y. Sh., Dushkin, M. I., and Kuznetsova,
I. S. (2000) J. Endotoxin Res., 6, 113-114.
14.Schwartz, Y. Sh., and Dushkin, M. I. (2001)
Ros. Kardiol. Zh., 30, 83-92.
15.Ulevitch, R. J. (1978) Immunochemistry,
15, 157-164.
16.Wood, F. T., Wu, M. M., and Gerhart, J. C.
(1975) Analyt. Biochem., 69, 339-349.
17.Lindgren, F. T. (1975) in Analysis of Lipids
and Lipoproteins (Perkins, E. G., ed.) American Oil Chemists
Society, Compaign, SL, USA, pp. 204-223.
18.Lowry, O. H., Rosebrough, N. J., Farr, A. L., and
Randall, R. J. (1951) J. Biol. Chem., 193, 265-275.
19.Glomset, J. (1968) J. Lipid Res.,
9, 155-167.
20.Vreugdenhil, A. C., Snoek, A. M., van 't Veer,
C., Greve, J. W., and Buurman, W. A. (2001) J. Clin. Invest.,
107, 225-234.
21.Niemann-Jonsson, A., Dimayuga, P., Jovinge, S.,
Calara, F., Ares, M. P., Fredrikson, G. N., and Nilsson, J. (2000)
Arterioscler. Thromb. Vasc. Biol., 20, 2205-2211.
22.Lopes-Virella, M. F. (1993) Eur. Heart J.,
14 (Suppl. K), 118-124.
23.Hamsten, A., Bjorkegren, J., Boquist, S.,
Nilsson, L., Ruotolo, G., Eriksson, P., Silveira, A., and Karpe, F.
(1998) in Atherosclerosis XI (Jacotot, B., Mathe, D., and
Fruchart, J.-C., eds.) Elsevier Science (Singapore) Pte Ltd., pp.
141-149.
24.Faergeman, O. (1998) in Atherosclerosis XI
(Jacotot, B., Mathe, D., and Fruchart, J.-C., eds.) Elsevier Science
(Singapore) Pte Ltd., pp. 605-607.
25.Packard, C. J., and Shepherd, J. (1998) in
Atherosclerosis XI (Jacotot, B., Mathe, D., and Fruchart, J.-C.,
eds.) Elsevier Science (Singapore) Pte Ltd., pp. 789-793.
26.lkeda, Y., Ashida, Y., Takagi, A., Fukuoka, T.,
Tsuru, A., Tsushima, M., and Yamomoto, A. (1998) in Atherosclerosis
XI (Jacotot, B., Mathe, D., and Fruchart, J.-C., eds.) Elsevier
Science (Singapore) Pte Ltd., pp. 777-788.
27.Victorov, A. V., Gladkaya, E. M., and Yurkiv, V.
A. (1989) Biokhimiya, 54, 434-443.
28.Victorov, A. V., Medvedeva, N. V., Gladkaya, E.
M., Morozkin, A. D., Podrez, E. A., Kosykh, V. A., and Yurkiv, V. A.
(1989) Biochim. Biophys. Acta, 984, 119-127.
29.Berger, D., Schleich, S., Seidelmann, M.,
and Beger, H. G. (1990) FEBS Lett.,277, 33-36.
30.Emancipator, K., Csako, G., and Elin, R. J.
(1992) Infect. Immun., 60, 596-601.
31.Wurfel, M. M., and Wright, D. (1995) in
Bacterial Endotoxins: Lipopolysaccharides from Genes to Therapy
(Levin, J., Alving, C. R., Munford, R. S., and Redl, H., eds.)
Wiley-Liss Inc., pp. 287-295.
32.Liao, W., and Floren, C. H. (1993) Scand. J.
Gastroenterol., 28, 97-103.
33.Hardardottir, I., Grunfeld, C., and Feingold, K.
R. (1994) Curr. Opin. Lipidol., 5, 207-215.
34.Auerbach, B. J., and Parks, J. S. (1989) J.
Biol. Chem., 264, 10264-10270.
35.Klimov, A. N., and Nikul'cheva, N. G. (1999)
Metabolism of Lipids and Lipoproteins and Its Disorders. Manual for
the Physicians [in Russian],3rd ed., Piter Kom, St. Petersburg.
36.Seguret-Mace, S., Benoit, P., Emmanuel, F.,
Caillaud, J. M., Bassinet, L., Viry, I., Gallix, P., Branellec, D.,
Denefle, P., and Duverger, N. (1998) in Atherosclerosis XI
(Jacotot, B., Mathe, D., and Fruchart, J.-C., eds.) Elsevier Science
(Singapore) Pte Ltd., pp. 1001-1010.