Effects of the Inhibitors of Dynamics of Cytoskeletal Structures on the Development of Apoptosis Induced by the Tumor Necrosis Factor
L. V. Domnina*, O. Yu. Ivanova, B. V. Cherniak, V. P. Skulachev, and J. M. Vasiliev
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: Domnina@mbio.genebee.msu.su* To whom correspondence should be addressed.
Received December 19, 2001; Revision received March 12, 2002
Changes in cytoskeletal structures have been investigated during apoptosis of epithelial HeLa cells induced by tumor necrosis factor-alpha (TNF-alpha). Shape and surface cell activity were investigated by time-lapse video microscopy, and changes of the cytoskeletal structure were studied by immune fluorescent microscopy. Addition of TNF-alpha to HeLa cell culture caused early disruption of the actin cytoskeleton and vinculin-containing focal contacts, keratin filaments, and microtubules. Rounding of cells, general blebbing, and nuclear fragmentation were observed at the terminal apoptotic stages. Actomyosin complex inhibitors, H7 and HA1077, suppressed blebbing (but not cell rounding) and activated the development of apoptosis. The latter suggests that in contrast to blebbing the general rounding does not depend on increased contractility of actomyosin cortex. These cytoskeletal inhibitors accelerated the development of apoptosis of HeLa cells and increased sensitivity of HeLa-Bcl-2 cells (transfected with DNA encoding antiapoptotic protein Bcl-2) to TNF-induced apoptosis. Damage of cytoskeletal structures significantly attenuated antiapoptotic activity of Bcl-2 in the HeLa-Bcl-2 cells. It is suggested that the stimulation of apoptosis by cytoskeletal inhibitors may be attributed to the altered distribution of cell organelles, especially, mitochondria.
KEY WORDS: TNF, apoptosis, actin, cytoskeleton, microtubules, vinculin, blebbing, mitochondria
Apoptosis, a programmed cell death, is realized via cascade activation of caspases (cysteine proteinases). It is characterized by a number of characteristic changes such as DNA fragmentation, condensation of nucleus and cytoplasm, and change of cell shape; the latter includes rounding and appearance of numerous evaginations of the cell surface known as blebbing [1-5]. These changes in the cell shape are obviously related to alterations of dynamic structure of a contractile submembrane layer, the actomyosin cortex. Recently, it was demonstrated that membrane blebbing stems from the caspase cleavage of Rho-associated kinase RhoI (ROCKI), a key enzyme regulating contractility of the actomyosin cortex. This cleavage results in constitutive activation of ROCKI [6-8]. However, a possible role of cytoskeleton changes in the development of the apoptotic scenario of cell death remains unclear. The central unanswered question is whether cytoskeletal changes represent secondary symptoms of terminal, so-called “executive” phase of the cell death, or they are important for the main manifestations of apoptosis, such as nuclear fragmentation.
In the present study we have analyzed the possible role of cytoskeleton in apoptosis by using two groups of inhibitors. These included inhibitors of kinases (including ROCKI) regulating contractility of the actomyosin cortex and inhibitors of actin polymerization, which are responsible for direct disruption of this cortex. We have also investigated the effect of compounds selectively damaging dynamic structure of microtubules, the other main component of cytoskeleton, on apoptosis.
In these studies we employed the standard model for induction of apoptosis: the effect of tumor necrosis factor (TNF) on HeLa cells and cells HeLa-Bcl-2 transfected with construct carrying DNA of antiapoptotic protein, Bcl-2. (HeLa-Bcl-2 cells are insensitive to TNF.)
TNF binding to its receptors causes rearrangement of adapter proteins and procaspases, particularly procaspase 8. Aggregation of procaspase 8 molecules is sufficient for formation of active caspase 8 which subsequently activates “executing” caspases 3, 6, and 7 [1, 9]. Mitochondria play a very important role in the development of apoptosis. Active caspase 8 cleaves Bid protein; its active form (tBid) is involved into damage to the outer mitochondrial membrane. The latter results in release of cytochrome c, apoptosis inducing factor (AIF), and some other mitochondrial proteins involved in the apoptotic cascade [5]. Bcl-2 and other related proteins represent antiapoptotic defense; they are localized in cytoplasm and mitochondrial membranes [2, 4, 10-12]. Using vital staining of mitochondria with rhodamine 123, we have also investigated changes in mitochondria redistribution in TNF-induced apoptotic cells.
MATERIALS AND METHODS
Cell cultures and light microscopy. Cell cultures HeLa and HeLa-Bcl-2 kindly presented by G. A. Belov (Institute of Poliomyelitis and Viral Encephalites, Russian Academy of Medical Sciences) were grown in Eagle medium containing 10% embryonal serum and antibiotics at 37°C and 5% CO2. Cell line HeLa-Bcl-2 was obtained by transfection of HeLa cells with vector pLPS-bcl-2. This vector contained the gene for puromycin resistance under control of LTR of Moloni mouse leucosis virus. This allowed selection of the transfected cells using medium containing puromycin (1 µg/ml). The difference between HeLa and HeLa-Bcl-2 cells was demonstrated by employing antibodies against Bcl-2 and Western-blot analysis. Vital dynamic observation of cell cultures was monitored using a video camera with time-lapse videotaping (Hamamatsu C2400, Japan). Cells were grown on cover glasses in special chamber with parallel walls [13]. The development of programmed cell death was observed after combined addition of 5 ng/ml TNF-alpha (which was kindly presented by V. G. Korobko, Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry) and protein synthesis inhibitor, 1 µg/ml emetine (Sigma, USA). TNF-induced apoptosis of HeLa cells requires inhibition of protein synthesis [14]. The role of cytoskeletal structures during cell entry to apoptosis induced by TNF and emetine was analyzed by adding 0.5 µg/ml colcemid (Sigma) and 10 µM taxol (Sigma) for microtubule damage, 1 µg/ml cytochalasin D (Sigma) and 2 µM latrunculin A (Molecular Probes, USA) for disruption of actin cytoskeleton structures, 200-500 µM H7 (Sigma) and 50 µM HA1077 (Alexis, USA) for inhibition of actomyosin contractility due to inhibition of myosin light chain phosphorylation. (HA1077 is a Rho kinase inhibitor [15], whereas H7 inhibits a wide range of kinases [16].)
Immunomorphology. For simultaneous registration of (co-)localization of actin and vinculin and actin and myosin II cells were fixed with 4% formaldehyde in phosphate buffer and extracted with 0.1% Triton X-100 in phosphate buffer for 3 min. Actin microfilament bundles were then stained with rhodamine-phalloidin (Sigma). Focal contacts, myosin II, and keratin filaments H1 were stained using polyclonal antibodies to vinculin (Sigma), non-muscular myosin (Biomedical Technologies, USA), and monoclonal antibodies to keratin 8, respectively. (Antibodies to keratin 8 were kindly presented by V. I. Gelstein and T. A. Chipysheva, Oncological Research Center, Russian Academy of Medical Sciences [17].) Microtubuli were stained with monoclonal antibodies to tubulin (DM1A, Sigma). FITC-conjugated anti-rabbit or anti-mouse IgG (Sigma) were used as secondary antibodies. Fragmentation of nuclei was determined by cell staining with fluorescent dye Hoechst 33342 (Sigma) and percent of apoptotic nuclei was presented as mean ± SD for 500 cells in each of three independent experiments. Statistical difference was evaluated by t-test, and results were considered as statistically significant at p < 0.05.
Vital staining of cells (used for studies of mitochondrial changes) with rhodamine 123 (0.5 µg/ml, Sigma) was carried out for 15 min.
Cells were investigated and photographed using an Axiophot microscope (Carl Zeiss, Germany) and objectives (Apochromat, Germany) of 40- and 60-fold magnification. Photo images from videotapes were obtained using the software Miro Video Capture DC10 (Germany).
RESULTS
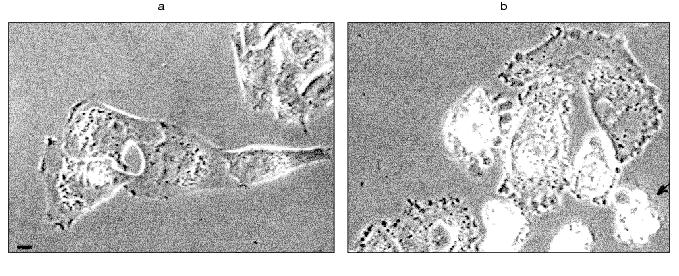
Dynamics of HeLa cell apoptosis. Videotaping of cell cultures revealed that the marginal blebbing was the earliest change of HeLa morphology: the appearance of short-living blebs over a cell perimeter was already noted 2 h after TNF addition. Sharp changes of the cell shape, such as rounding and blebbing appeared later (from 3 to 4 h after addition of TNF and emetine). However, they did not appear simultaneously: initially a cell remained spreading, then it rapidly (within 10 sec) contracted. Blebbing of the whole surface of the rounding cell lasted for 1-2 h (Fig. 1, a and b). Fragmentation of nuclei (Hoechct 3342 staining) was noted only in rounding cells. After 7 h of TNF this was noted in 70% cells (Fig. 2, e and f). Apoptosis of all cells (100%) was found after 24 h.
Fig. 1. Bleb formation in HeLa cells induced by TNF and emetine. Vital videotaping of HeLa cells. Phase contrast: a) control; b) treatment with TNF + emetine for 2 h. Rounding is noted in five cells, arrow indicates a cell with general blebbing. In all figures the scale of strip is 10 µm.
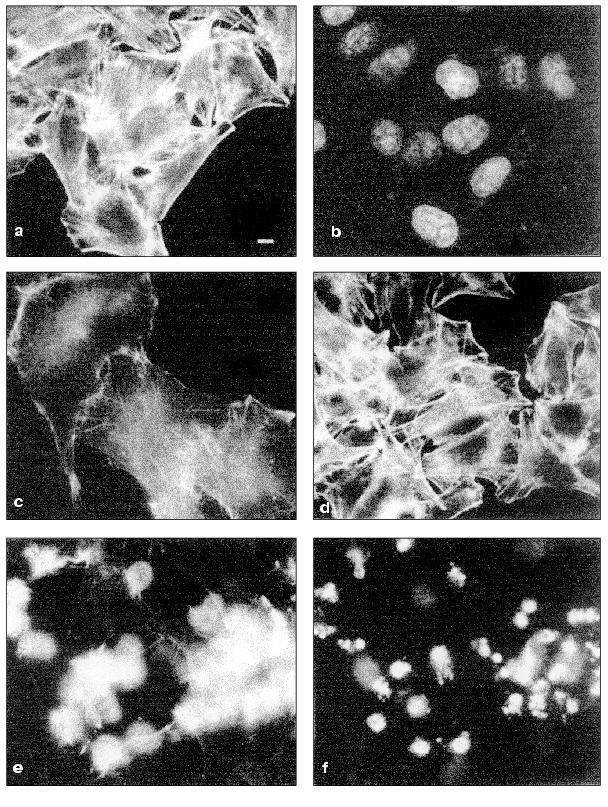
Changes of cytoskeleton. Table summarizes TNF-induced changes in cytoskeletal structures of HeLa cells.Fig. 2. Redistribution of actin microfilament bundles in HeLa and HeLa-Bcl-2 cells induced by TNF + emetine. Cells grown for 2 days were stained for actin (a, c, d, e) and DNA (b, f). a, b) HeLa cells before administration TNF and emetine; actin bundles are visible in cytoplasm (a) no fragmented nuclei (b); c) the same cells treated with TNF and emetine for 2 h; actin bundles disappeared; d) HeLa-Bcl-2 cells treated with TNF and emetine for 7 h; cells are spread and bundles are well defined; e, f) HeLa cells treated with TNF and emetine for 7 h; cell rounding (e) and fragmentation of nuclei (f) are visible; a-c, e, f) HeLa; d) HeLa-Bcl-2.
Changes in cytoskeletal structure in spreading HeLa cells at various
time intervals after addition of TNF and emetine
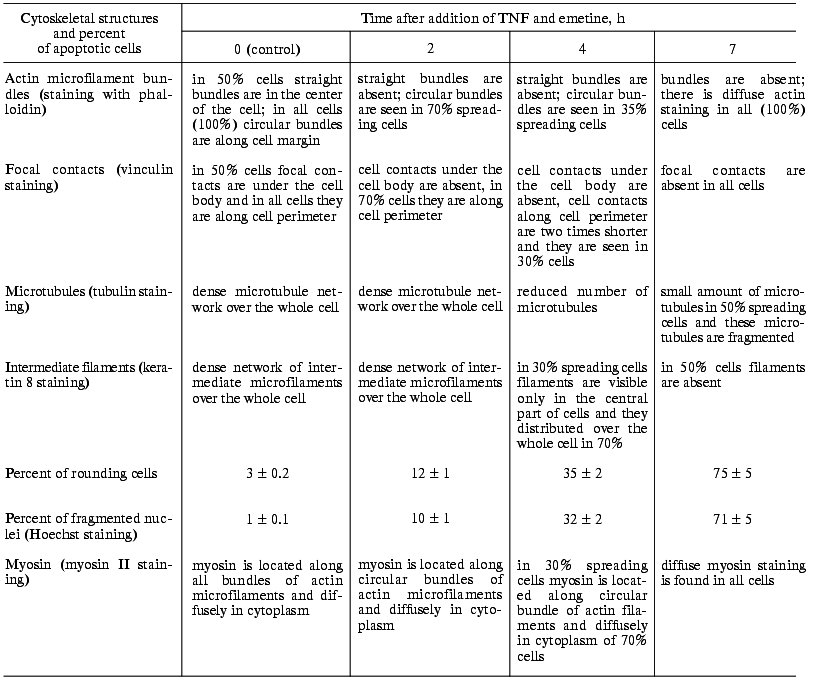
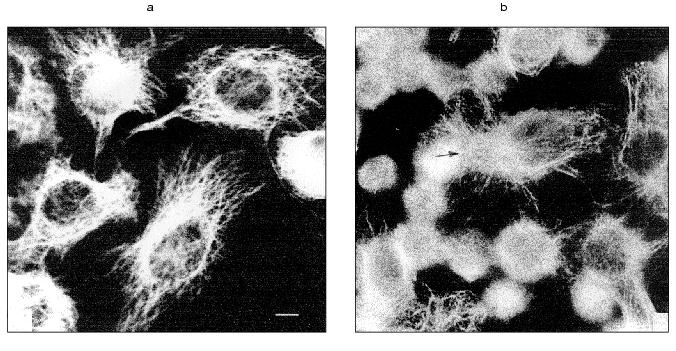
In spreading cells TNF treatment for 2 h caused disruption of actin microfilament bundles localized in cytoplasm and myosin II structures associated with these actin microfilaments. With prolonged TNF treatment (for 6-7 h) circular microfilament bundles disappeared in almost all cells (Fig. 2, a and c) and myosin staining became diffuse. Disruption of actin microfilament bundles was accompanied by reduction of vinculin-positive focal contacts (initially under cell body and then on periphery of the cell) (Fig. 3). The keratin filament network was also disrupted: after 4-7 h of TNF effect filaments of many cells shortened, fragmented, and disappeared from lamelloplasma (Fig. 4). The third main cytoskeletal system, microtubules, was more resistant to TNF treatment. Nevertheless, in some cells the microtubular network become sparser and the number of microtubules was reduced (Fig. 5).
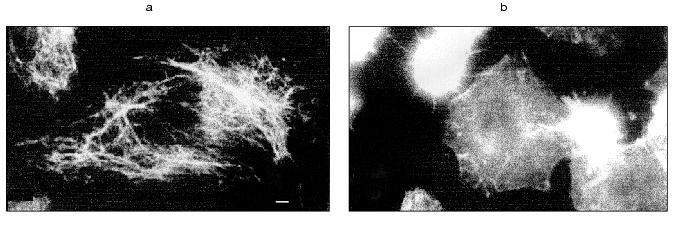
Fig. 3. Redistribution of vinculin-positive focal contacts (indicated by arrows) in HeLa cells treated with TNF and emetine. Cells grown for 2 days were stained for vinculin; before (a) and after treatment with TNF and emetine for 4 h (b); after the treatment number of focal contacts on the cell margin is sharply reduced.
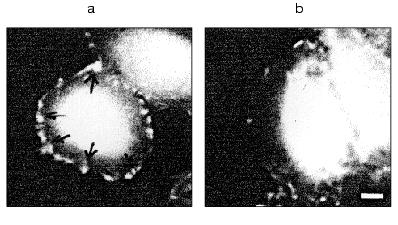
Fig. 4. Redistribution of keratin fibers in HeLa cells treated with TNF and emetine. Cells grown for 2 days were stained for keratin; before (a) and after treatment with TNF and emetine for 4 h (b); after the treatment the number of keratin filaments is sharply reduced in the spreading cell.
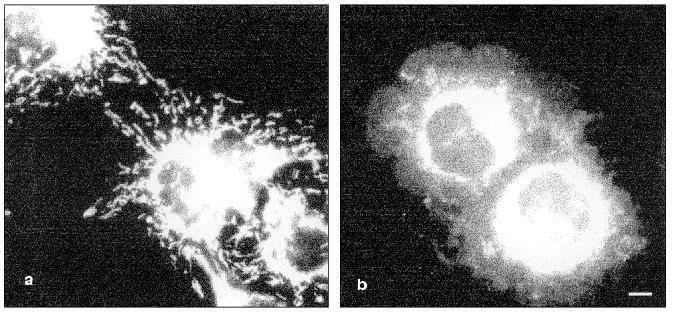
TNF treatment (for 4-6 h) also influenced the intracellular distribution of mitochondria: in contrast to control cells where mitochondria were localized over all the cytoplasm, in TNF-treated cells they concentrated around the nucleus (Fig. 6).Fig. 5. Redistribution of microtubules in HeLa cells treated with TNF and emetine. Cells grown for 2 days were stained for tubulin; before (a) and after treatment with TNF and emetine for 4 h (b); the treatment resulted in reduction of microtubule network in the spreading cells (arrow).
Effect of cytoskeletal inhibitors on HeLa cells. The kinase inhibitors HA1077 and H7 (which reduce contractility of actomyosin complex) did not induce apoptosis in HeLa cells over 24 h treatment. During their addition together with TNF, actomyosin microfilament bundles disappeared after 2-4 h and diffusely stained actin was noted in the cytoplasm. The onset and frequency of cell rounding did not differ from those in cells treated with TNF. However, marginal blebs on the surface of spreading cells were absent. The combined treatment of HeLa cells with HA1077 (or H7) with TNF shortened general blebbing from 1-2 to 0.5 h and lifetime of blebs reduced to a fraction of a second. The combined treatment of cells with TNF and the kinase inhibitors for 4 and 7 h also increased percent of fragmented nuclei compared with effect of TNF alone (Fig. 7).Fig. 6. Vital staining of mitochondria with rhodamine 123 before (a) and after the treatment of cells with TNF and emetine for 4 h (b). After the treatment mitochondria are localized in the central part of the cytoplasm.
The presence of cytochalasin D resulted in disruption of actin cytoskeleton and disappearance of microfilament bundles followed by appearance of point accumulations of actin. Cytochalasin D itself did not induced apoptosis in HeLa cells over 24 h. However the combined treatment with TNF and cytochalasin D increased the number of cells with fragmented nuclei compared with the effect of TNF alone (Fig. 8).Fig. 7. Histogram of the effects of various agents influencing actomyosin cytoskeleton on TNF-induced apoptosis in HeLa cells after 4 h (light rectangles) and 7 h (dark rectangles). EM, emetine; CCD, cytochalasin D.
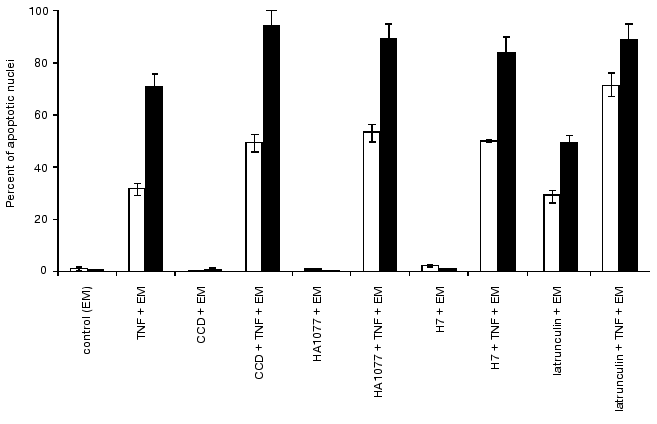
Addition of latrunculin A caused rapid (within 10 min) disruption of actin microfilaments, which was accompanied by appearance of diffusely stained actin. Latrunculin A itself induced appearance of rounding cells with fragmented nuclei; after 7 h the percent of latrunculin A-treated cells (50%) with fragmented nuclei was comparable with the value observed during TNF-induced apoptosis (56%) (Fig. 8). During combined treatment of cells with latrunculin A and TNF for 7 h the percent of cells with fragmented nuclei reached 90%.Fig. 8. Histogram of the effects of various agents disrupting microtubules on TNF-induced apoptosis in HeLa cells after 4 h (light rectangles) and 7 h (dark rectangles).
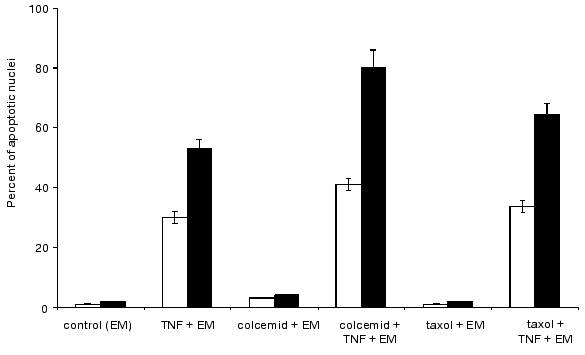
Colcemid and taxol influencing cellular system of microtubules did not induce apoptosis over 24 h. However, during combined treatment of cells with colcemid (or taxol) and TNF for 7 h the percent HeLa cells with fragmented nuclei was higher than after the same treatment with TNF (Fig. 8).
Effect of cytoskeletal inhibitors on HeLa-Bcl-2 cells. Morphology and cytoskeletal structure of HeLa-Bcl-2 cells were similar to those of HeLa cells. However, treatment of these cells with TNF for 7 h had no effect on cytoskeletal structures (Fig. 2d). In contrast to HeLa cells after 7 h treatment with TNF rounding and fragmentation of nuclei were found only in 10-15% of HeLa-Bcl-2 cells.
The cytoskeletal inhibitors HA1077, H7, cytochalasin D, taxol, and colcemid did not induce apoptosis during cell treatment for 7 or 24 h. Only the treatment with latrunculin A for 7 h slightly increased the number of cells with fragmented nuclei (Fig. 9).
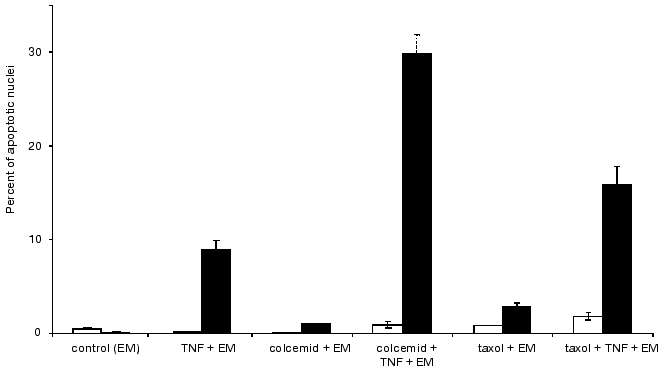
Cytochalasin D and latrunculin A, colcemid, taxol, and HA1077 sharply increased percent of rounding apoptotic cells in TNF-treated HeLa-Bcl-2 cultures; after 7 h it was from two to three times higher than in the cells treated only with TNF but significantly lower than in non-transfected HeLa cells treated in the same way (Figs. 9 and 10).Fig. 9. Histogram of the effects of various agents influencing actomyosin cytoskeleton on TNF-induced apoptosis in HeLa-Bcl-2 cells after 4 h (light rectangles) and 7 h (dark rectangles).
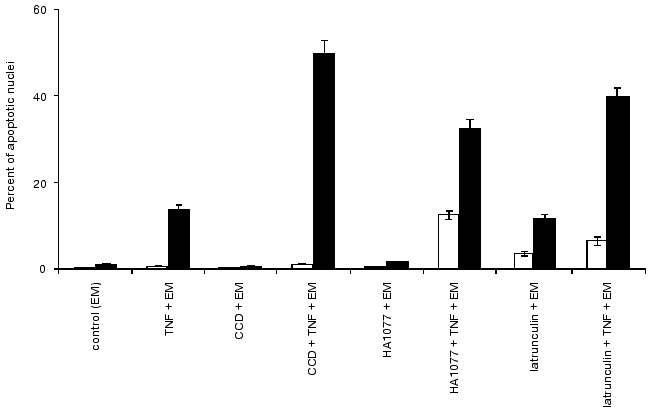
Fig. 10. Histogram of the effects of various agents disrupting microtubules on TNF-induced apoptosis in HeLa-Bcl-2 cells after 4 h (light rectangles) and 7 h (dark rectangles).
DISCUSSION
The apoptotic process is accompanied by destructive changes in all types of cytoskeletal structures: actin cortex, microtubules, and intermediate filaments. Disruption of actomyosin bundles obviously represents the earliest visible changes. Changes of actomyosin cortex are probably responsible for the manifestation of the last stage of apoptosis, blebbing (formation of multiple contractile blebs) of the cell surface. Our experiments with inhibitors of actomyosin cortex contractility support conclusions of other authors [6-8] that blebbing is the result of stimulation of such contractility. It is possible that this stimulation represents a consequence of caspase-induced irreversible activation of one of the key enzymes, regulating cortex contractility, serine-threonine ROCKI kinase [6, 7]. According to our data inhibitors of actomyosin contractility suppress blebbing, but they do not influence cell rounding, another symptom of the terminal phase of apoptosis. This suggests that the rounding is not related to the stimulation of cortex contractility and involves some other mechanism (e.g., rounding may be related to caspase mediated disruption of the system focal contacts-integrins-matrix [18, 19]). Moreover, blebbing inhibitors even increased fragmentation of nuclei, the most typical manifestation of apoptosis. The latter suggests that bleb formation is not required for the development of destructive changes in the nucleus. Perhaps, bleb formation might be considered as a protective reaction, attenuating general cell destruction: e.g., blebs could be responsible for sequestration of some cell components involved into realization of apoptotic program. Some evidence exists that caspases are accumulated in apoptotic blebs [20].
In our experiments almost all agents damaging cytoskeleton integrity (cytochalasin, latrunculin, colcemid) accelerated TNF-induced apoptosis. It was previously shown that many of these inhibitors (e.g., cytochalasin and colcemid [21, 22]) may induce apoptosis themselves. In the present study we have found that latrunculin, the agent causing selective reversible depolymerization of actin microfilaments [23, 24], may induce apoptosis.
Cytoskeleton damages caused in our experiments by various agents were able to overcome protein Bcl-2 protection against TNF-induced apoptosis. Comparison of kinetics of cytoskeletal disruptions and manifestations of “marker” apoptotic events (such as DNA fragmentation) is methodologically difficult task because of asynchronous development of these events in the cell culture.
The most characteristic effect of Bcl-2 consists of prevention of cytochrome c release from mitochondria into cytoplasm; this release causes activation of caspase 3 and plays a key role in the development of later manifestations of apoptosis [1, 12, 24]. However, in our experiments Bcl-2 also exerted a protective effect on relatively early signs of apoptosis (e.g., disruption of actin filament bundles).
Thus, data presented here clearly demonstrate that various damages to normal cytoskeletal dynamics, such as attenuation of actomyosin cortex contractility or disruption of the structure of microtubules, can not only induce apoptosis but also stimulate programmed cell death induced by the activation of specific receptor.
What are the possible mechanisms of the proapoptotic effects of cytoskeletal changes? We believe that cytoskeleton-dependent changes in positioning and functioning of mitochondria may be involved in these mechanisms. In fact, mitochondria play a crucial role in the dynamics of apoptosis. It is also known that one of the main functions of cytoskeleton consists of control of positioning and movement of intracellular organelles including mitochondria [5, 25, 26].
Changes in distribution of mitochondria are actually observed during apoptosis. In previous [27, 28] and the present studies (Fig. 6) it was demonstrated that in TNF-treated cells mitochondria left peripheral zones of cytoplasm and accumulated in the perinuclear central zone. It is possible that such redistribution results from inhibition of functioning of kinesin, a molecular motor responsible for organelle movement along microtubules to the periphery. Disruption of microtubules by colcemid is also accompanied by mitochondria accumulation in the central zone of the cell [26]. It is possible that such changes in the positioning of mitochondria can stimulate release of cytochrome c and other proteins into the central zone and therefore potentiate destructive changes in nuclear structures.
This and other hypotheses on the interrelationship between mitochondrial and cytoskeletal changes in apoptosis require further experimental investigation.
Authors are very grateful to O. Yu. Pletjushkina (Belozersky Institute of Physico-Chemical Biology) for constructive discussion, G. A. Belov (Institute of Poliomyelitis and Viral Encephalites) for generous gift of HeLa and HeLa-Bcl-2 cell cultures, and V. G. Korobko (Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry) for supply of TNF.
This work was supported by grants from the Ludwig Institute for Cancer Research (PO 863) and the Russian Foundation for Basic Research (Nos. 99-04-49256, 99-04-49126, 00-04-48090).
REFERENCES
1.Kopnin, B. P. (2000) Biochemistry (Moscow),
65, 2-27.
2.Metcalfe, A., and Streuli, Ch. (1997) Bio
Essays,19,711-720.
3.Mills, J. C., Stone, N. L., Erhardt, J., and
Pittman, R. N. (1998) J. Cell Biol.,140,627-636.
4.Pawlak, G., and Helfman, D. M. (2001) Curr.
Opin. Genet. Devel.,11, 41-47.
5.Skulachev, V. P. (2001) Exp.
Gerontol.,36, 995-1024.
6.Coleman, M. L., Sahai, E. A., Yeo, M., Bosch, M.,
Dewar, A., and Olson, M. F. (2001) Nature Cell Biol., 3,
339-345.
7.Leverrier, Y., and Ridley, A. J. (2001) Nature
Cell Biol.,3,91-93.
8.Sebbagh, M., Renvoilz, C., Hamelin, J., Riche, N.,
Bertoglio, J., and Breagd, J. (2001) Nature Cell Biol.,
3, 346-352.
9.Chang, H. Y., and Yang, X. (2000) Microbiol.
Mol. Biol. Rev.,821-846.
10.Reed, J. C. (1994) J. Cell
Biol.,124, 1-6.
11.Reed, J. C. (1997) Nature,387,
773-776.
12.Reed, J. C. (1997) Adv. Pharmacol.,
41, 501-532.
13.Rovensky, Y. A., Domnina, L. V., Ivanova, O. Y.,
and Vasiliev, J. M. (1999) J. Cell Sci.,112,
1273-1282.
14.Sidoti-de Fraisse, C., Rincheval, V., Risler, Y.,
Mignotte, B., and Vayssiere, J.-L. (1998) Oncogene,17,
1639-1651.
15.Seto, M., Sasaki, Y., and Hidaka, H. (1991)
Eur. J. Pharmacol.,195, 267-272.
16.Cici, S. S., Volberg, T., Bershadsky, A. D.,
Denisenco, N., and Geiger, B. (1994) J. Cell Sci., 107,
683-692.
17.Guelshtein, V. I., Tchypisheva, T. A., Ermilova,
V. D., Litvinova, L. V., Troyanovsky, S. M., and Bannikov, G. A.
(1988) Int. J. Cancer,42, 147-153.
18.Bratton, S. B., and Cohen, G. M. (2001) Trends
Pharmacol. Sci.,22, 306-315.
19.Stegh, A. H., Herrmann, H., Lampel, S.,
Weisenberger, D., Andra, K., Seper, M., Wiche, G., Krammer, P. H., and
Peter, M. E. (2000) Mol. Cell Biol.,20, 5665-5679.
20.Packard, B. Z., Komoriya, A., Brots, T. M., and
Henkart, P. A. (2001) Immunology,167, 5061-5066.
21.Rubtsova, S. N., Kondratov, R. V., Kopnin, P. B.,
Chumakov, P. M., and Vasiliev, J. M. (1998) FEBS Lett.,
430, 353-357.
22.Volbracht, C., Leist, M., and Nicotera, P.
(1999) Mol. Med.,5, 477-489.
23.Spector, I., Shochet, N. R., Kashman, Y., and
Groweiss, A. (1983) Science, 219, 493-495.
24.Spector, I., Shochet, N. R., Blasberger, D., and
Kashman,Y. (1989) Cell. Motil. Cytoskeleton, 13,
127-144.
25.Constantini, P., Jacotot, E., Decaudin, D., and
Kroemer, G. (2000) Nat. Cancer Inst., 92, 1042-1053.
26.Radionov, V. I., Gyoeva, F. K., Tanaka, E.,
Bershadsky, A. D., Vasiliev, J. M., and Gelfand, I. M. (1993) J.
Cell. Biol., 123, 1811-1820.
27.De Vos, K., Goossens, V., Boone, E., Vezcammen,
D., Vancompernoll, K., Vendenabeele, P., Haegeman, G., Fiers, W., and
Grooten, J. (1998) J. Biol. Chem., 273, 9673-9680.
28.De Vos, K., Severin, F., van Herrewoghe, F.,
Gossens, V., Hyman, A., and Grooten, J. (2000) J. Cell Biol.,
149, 1207-1214.