DNA Aptamers as Radically New Recognition Elements for Biosensors
V. A. Spiridonova1* and A. M. Kopylov1,2
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119992 Russia; fax: (095) 939-3181; E-mail: spiridon@genebee.msu.su2School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia
* To whom correspondence should be addressed.
Received November 1, 2001
A fiber-optic biosensor based on DNA aptamers used as receptors was developed for the measurement of thrombin concentration. Anti-thrombin DNA aptamers were immobilized on silica microspheres, placed inside microwells on the distal tip on an imaging optical fiber, coupled to a modified epifluorescence microscope through its proximal tip. Thrombin concentration is determined by a competitive binding assay using a fluorescein-labeled competitor. The biosensor is selective and can be reused without any sensitivity change. The thrombin limit of detection is 1 nM, sample volume is 10 µl, and assay time per sample is 15 min including the regeneration step.
KEY WORDS: DNA aptamers, thrombin, glass fiber, biosensor, SELEX
The practical application of fundamental knowledge about DNA has been rapidly grown over the last decade. A development of high technology analytical methods based on immobilized DNA is now an effectively progressing field. The main achievement of microarray technology, so-called DNA-chips [1-3], is the ability to use the diversity of DNA libraries, amplified by polymerase chain reaction (PCR) to produce DNA probes for rapid analysis of thousands of genes and their mutant forms [4, 5], DNA polymorphism [6], for gene discovery and monitoring of gene expression [7-9], i.e., to perform a multiple genome microanalysis using a hybridization method.
DNA-chips are based on DNA fragments with a length of tens to hundreds of nucleotides, immobilized on an activated solid support1. Using the PIN-micromanipulator the robot transfers less than 1 nl of DNA solution from the microtiter plate well onto a corresponding microchip cell, with a droplet diameter of 150 µm. This procedure is performed by a robot, and some thousands of DNA probes can be deposited on a support of a few cm2. Thus, every cell contains a certain part of the genetic information, and the integration of the DNA microchips within a single platform allows construction of a chip array with a full package of the genetic information, available for any identification purpose [Footnote: For more details see www.biochip.ru].
The development of microarrays based on DNA aptamers used as receptors can be seen as a logical continuation of the DNA-chip technology development, although the principle of target recognition is not based on hybridization, but is analogous to the immunochemical assay. Aptamers are antibody analogs in terms of both specificity and affinity, with an apparent advantage of the former to be reproduced by automated chemical synthesis.
Aptamers are single-stranded oligonucleotides with a length of tens of nucleotides, obtained by SELEX technology, and exhibiting high affinity and specificity towards any defined recognition target. Aptamers are selected by SELEX technology (Systematic Evolution of Ligands by Exponential enrichment), which is discussed in detail in a number of reviews [10-22] [Footnote: The first issue of Current Opinion in Chemical Biology (1997) is focused on combinatorial chemistry including SELEX (see [19])].
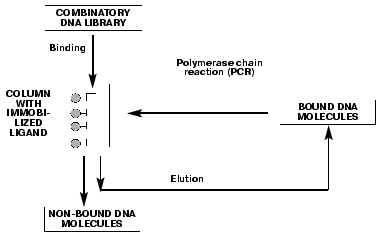
Single-stranded DNA aptamers have a very highly ordered tertiary structure, which allows them to form stable and specific complexes with different targets such as proteins, nucleic acids, low molecular weight compounds, etc. The search for the aptamers begins with a selection of “random sequence” DNA library. Libraries are obtained by automated DNA synthesis with a gradient attachment of synthons using a mixture of nucleotides instead of single components. Usually, a randomized region is 30 to 60 nucleotides, flanked on both sides with a special DNA sequence for PCR amplification. The diversity of individual oligonucleotides in the random DNA libraries can be as high as of 1015-1018 molecules. A principle of SELEX technology for DNA aptamers, consisting of several consecutive selection cycles, is shown in Fig. 1. The key step is DNA binding with an immobilized ligand, e.g., protein. During this step, oligonucleotides with an affinity towards the target compound are selected from the tremendous number of molecules in the library. Unbound DNA molecules are eluted from the column, while bound aptamers are isolated from the complex. DNA aptamers are amplified by PCR, and a single chain DNA is obtained from the resulting DNA duplex using an asymmetric PCR, and then is used for a next selection cycle. Aptamers with a low affinity and specificity towards the target are displaced by that ones with high affinity and specificity in the following selection cycles. Basically, the DNA fraction obtained after several selection cycles is a mixture of aptamers with various affinities towards the target, but individual aptamers can be isolated by cloning and sequencing of the fraction. DNA molecules matching by the primary structure are identified among the isolated aptamers and used for the quantitative estimation of the aptamer affinity for the target. The stability of the complexes is characterized by the apparent dissociation constants (Kd). These values for the aptamer-protein complexes vary within the 1-100 nM range, which is of similar affinity and specificity range as for antibody-antigen complexes.
DNA aptamers towards the serine protease thrombin, which is a key enzyme involved in the blood coagulation mechanism [23], were obtained [24-26] using SELEX technology. It should be mentioned that since thrombin is not a DNA-binding protein, the possibility to elicit thrombin-aptamer complexes clearly demonstrates the power of SELEX technology.Fig. 1. The principle of SELEX technology for DNA aptamers is based on a number of repetitive binding cycles of the DNA library containing a random sequence to a target compound.
Bock et al. [24] have obtained a family of DNA aptamers capable of inhibiting thrombus formation in vitro. Primary structures of all isolated aptamers contained a unique pentanucleotide motif with a characteristic distribution of guanine residues, engaged in formation of G-quartet structures similar to those found at the ends of telomeric DNA (PDB 156D). Macaya et al. [25] and Tsiang et al. [26] (see also [27, 28]) obtained other thrombin-specific DNA aptamers that had more sophisticated architecture of G-quartets with duplexes and additional hairpin loops and were able to form more stable complexes with thrombin. Values of apparent dissociation constants for thrombin-aptamer complexes were within the nM to µM range.
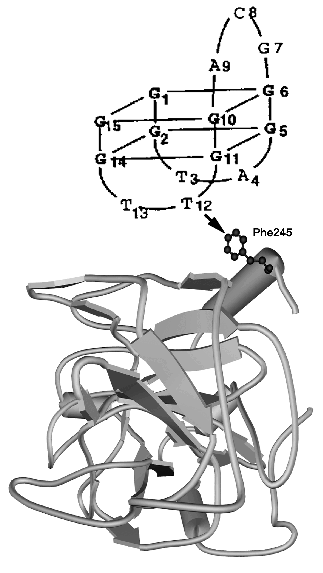
The tertiary structure of thrombin alone was determined by X-ray analysis (PDB 1JOU). The structure of DNA aptamer was determined by NMR, revealing two G-tetrads connected through two TT and one TGT loops (PDB 148D, see also [28]). The model of thrombin-aptamer complex was based on DNA-protein cross-linking data (Fig. 2).
Since they were first discovered, DNA aptamers have been considered for potential use as therapeutic agents [20, 29, 30], and only recently they also have been adopted as new recognition elements for biosensors [31] and flow-cytometry (see references in [20]). The first report on this application was published by Lee and Walt from Tafts University USA [32]. They used anti-thrombin DNA aptamers provided by E. Ellington (Center for Aptamer Research, Indiana University, USA), one of the leaders in SELEX technology development. Aptamers were covalently immobilized on the surface of non-porous silica microspheres of 3 µm diameter, according to Ferguson et al. [33]. A six-carbon atom spacer with an active end amino group was used to modify the 5´-end of 15-mer DNA aptamer GGTTGGTGTGGTTGG, and the resulting derivative was activated by cyanuric chloride. Hydrophilic microspheres with surface silanol group (Si-OH) [34] displayed a low degree of nonspecific hydrophobic thrombin binding. Microspheres were first reacted with (3-aminopropyl)-triethoxysilane, then with glutaraldehyde and polyethylenimine (PEI). PEI ensures high bead capacity and provides a spacer arm, which in combination with six-carbon atom spacer enhances the steric accessibility of immobilized aptamer to thrombin.Fig. 2. Model of the tertiary structure for thrombin (PDB 1JOU)-aptamer DNA (PDB 148D) complex. An arrow shows the contact determined by the chemical cross-linking method [27].
Microbeads with immobilized DNA aptamers were placed into microwells (3 µm diameter) that were fabricated by etching of the distal tip of the imaging fiber (500 µm diameter) with hydrofluoric acid. Microwell volume was 10 µl. The proximal tip of the optical imaging fiber was coupled to a modified extension of epifluorescence microscope [35]. A solution of fluorescein-labeled thrombin (3000 NIH units/mg, Sigma, USA) was pipetted onto the microwells of the image fiber with embedded microspheres. Fluorescein-labeled thrombin (F-thrombin) was synthesized by a reaction of thrombin with fluorescein succinimidyl ester. F-Thrombin was selectively bound by the microspheres with immobilized aptamers, and light emitted by a fluorescent label was registered by the microscope. Every microbead could be identified with 90% precision. Though signals from the individual microspheres showed a significant dispersion, averaged signals from 100 microspheres ensured a reliable detection (RSD 3%) [32]. Apparent Kd for F-thrombin was 300 nM, this exceeding the previously reported values of 50-100 nM [25, 31]1. It might be due to the chemical modification of thrombin. Nonspecific binding of F-thrombin to the blank microspheres was below 5%. F-Thrombin limit of detection was 10 nM. Specificity of aptamer-coated beads for F-thrombin binding was assessed with bovine serum albumin [Footnote: According to our data, apparent Kd values for the aptamers of this type are substantially dependent on specific functional activity of a particular thrombin sample (Spiridonova et al. (2002) Biochemistry (Moscow), in press)].
The most convenient assay format of thrombin analysis for routine medical purposes is, however, a competitive binding assay. Optimal F-thrombin concentration for thrombin analysis was found to be 200 nM. The method exhibited good results over nM to µM thrombin concentration range, thus matching the thrombin physiological concentration range in blood plasma, which is within 5-500 NIH units/ml [36, 37]. Hence, the method can be applied for thrombin quantification in real blood plasma samples. Assay time per sample is 15 min including the regeneration step: thrombin binding by the aptamer microbeads was complete in 7-8 min, and the regeneration was performed in 5-6 min. No change of biosensor parameters was detected over 8 h operation time. The biosensor can be reused many times without loss of sensitivity. Storage stability of the microspheres with immobilized aptamers was over 3 months.
Competitive F-thrombin binding assay is analogous to traditional Enzyme Linked Immunosorbent Assay (ELISA) and can be performed using standard ELISA microtiter plates [38]. Moreover, the present investigation demonstrates new possibilities for multi-analyte detection, which is governed entirely by the affinity and specificity of the DNA aptamers [Footnote: While this manuscript was in preparation, several new interesting papers have been published [39-42]].
Authors would like to thank Professor S. M. Strukova for useful discussions and senior scientist I. N. Kurochkin for a careful reading of the manuscript.
Financial support from the projects funded by the Russian Foundation for Basic Research (grant Nos. 01-04-48603, 01-04-48643, 99-04-39072), Ministry of Science (grant No. 415/99), and Universities of Russia (grant No. 99-1700) is greatly acknowledged.
REFERENCES
1.Chee, M., Yang, R., Hubbell, E., Berno, A., Huang,
X. C., Stern, D., Winkler, J., Lockhart, D. J., Morris, M. S., and
Fodor, S. P. A. (1996) Science, 274, 610-614.
2.McGall, G., Labadie, J., Brock, P., Wallraff, G.,
Nguyen, T., and Hinsberg, W. (1996) Proc. Natl. Acad. Sci.
USA, 93, 13555-13560.
3.Marshall, A., and Hodgson, J. (1998) Nature
Biotechnol.,16, 27-31.
4.Shuber, A. P., Michalowsky, L. A., Nass, G. S.,
Skoletsky, J., Hire, L. M., Kotsopoulos, S. K., Phipps, M. F.,
Barberio, D. M., and Klinger, K. W. (1997) Hum. Mol. Genet.,
6, 337-347.
5.Hacia, J. G., Brody, L. C., Chee, M. S., Fodor, S.
P. A., and Collins, F. S. (1996) Nature Genet., 14,
441-447.
6.Wang, D. G., Fan, J., Siao, C., Berno, A., Young,
P., Sapolsky, R., Ghandour, G., Perkins, N., Winchester, E., Spencer,
J., Kruglyak, L., Stein, L., Hsie, L., Topaloglou, T., Hubbell, E.,
Robinson, E., Mittmann, M., Morris, M. S., Shen, N., Kilburn, D.,
Rioux, J., Nusbaum, C., Rozen, S., Hudson, T. J., Lipshutz, R., Chee,
M., and Lander, E. S. (1998) Science,280, 1077-1082.
7.Schena, M., Shalon, D., Heller, R., Chai, A.,
Brown, P. O., and Davis, R. W. (1996) Proc. Natl. Acad. Sci.
USA,93, 10614-10619.
8.DeRisi, J., Penland, L., Brown, P. O., Bittner, M.
L., Meltzer, P. S., Ray, M., Chen Y., Su, Y. A., and Trent, J. M.
(1996) Nature Genet., 14, 457-460.
9.De Saizieu, A., Certa, U., Warrington, J., Gray,
C., Keck, W., and Mous, J. (1998) Nature Biotechnol.,16,
45-48.
10.Burke, J. M., and Bezzal-Herranz, A. (1993)
FASEB J., 7, 106-112.
11.Klug, S. J., and Famulok, M. (1994) Mol. Biol.
Report, 20,97-107.
12.Breaker, R. R., and Joyce, G. F. (1994)
TIBTECH, 12, 268-275.
13.Gold, L., Polisky, B., Uhlenbeck, O., and Yarus,
M. (1995) Annu. Rev. Biochem., 64, 763-797.
14.Gold, L. (1995) J. Biol. Chem.,
270, 13581-13584.
15.Iverson, B. L. (1995) Chem. Biol.,
2,67-70.
16.Bartel, D. P., and Szostak, J. W. (1996) in
RNA-Protein Interactions (Nagai, K., and Mattaj, I. W., eds.)
IRL Press, Oxford-New York-Tokyo, pp. 248-268.
17.Eaton, B. E., Gold, L., Hicke, B. J., Janjic, N.,
Jucker, F. M., Sebesta, D. P., Tarasow, T. M., Willis, M. C., and
Zichi, D. A. (1997) Bioorg. Med. Chem., 5, 1087-1096.
18.Gold, L., Brown, D., He, Y.-Y., Shtatland, T.,
Singer, B. S., and Wu, Y. (1997) Proc. Natl. Acad. Sci. USA,
94, 59-64.
19.Osborne, S. E., Matsumura, I., and Ellington, A.
D. (1997) Curr. Opin. Chem. Biol., 1, 5-9.
20.Kopylov, A. M., Spiridonova, V. A., and Park,
K.-H. (1998) Ros. Khim. Zh.,42, 89-96.
21.Kopylov, A. M., and Spiridonova, V. A. (2000)
Mol. Biol. (Moscow), 34, 1097-1113.
22.Breaker, R. R. (1997) Curr. Opin. Chem.
Biol., 1,26-31.
23.Tsiang, M., Jain, A. K., Dunn, K. E., Rojas, M.
E., Leung, L. K., and Gibbs, C. S. (1995) J. Biol. Chem.,
270, 16854-16863.
24.Bock, L. C., Griffin, L. C., Latham, J. A.,
Vermaas, E. H., and Toole, J. J. (1992) Nature, 355,
564-566.
25.Macaya, R. F., Waldron, J. A., Beutel, B. A.,
Gao, H., Joesten, M. E., Yang, M. M., Patel, R., Bertelsen, A. H., and
Cook, A. F. (1995) Biochemistry, 34, 4478-4492.
26.Tsiang, M., Gibbs, C. S., Griffin, L. C., Dunn,
K. E., and Leung, L. K. (1995) J. Biol. Chem., 270,
19370-19376.
27.Tasset, D. M., Kubik, M. F., and Steiner, W.
(1997) J. Mol. Biol., 272, 688-698.
28.Schultze, P., Macaya, R. F., and Feigon, J.
(1994) J. Mol. Biol., 235, 1532-1545.
29.Jenison, T. D., Gill, S. C., Pardi, A., and
Polisky, B. (1994) Science, 263, 1425-1429.
30.Hicke, B. J., Watson, S. R., Koenig, A., Lynott,
C. K., Bargatze, R. F., Chang, Y., Ringquist, S., Moon-McDermott, L.,
Jennings, S., Fitzwater, T., Han, H., Varki, N., Albinana, I., Willis,
M. C., Varki, A., and Parma, D. (1996) J. Clin. Invest.,
98, 2688-2692.
31.Potyrailo, R. A., Conrad, R. C., Ellington, A.
D., and Hieftje, G. M. (1998) Analyt. Chem., 70,
3407-3412.
32.Lee, M., and Walt, D. R. (2000) Analyt.
Biochem., 282, 142-146.
33.Ferguson, J. A., Boles, T. C., Adams, C. P., and
Walt, D. R. (1996) Nature Biotechnol., 14, 1681-1684.
34.Lundstrom, I., Ivarsson, B., Jonsson, U., and
Elwing, H. (1987) in Polymer Surfaces and Interfaces
(Feast, W. J., and Munro, H. S., eds.) Wiley, New York, p. 201.
35.Bronk, K. S., Michael, K. L., Pantano, P., and
Walt, D. R. (1995) Analyt. Chem., 67, 2750-2757.
36.Fenton, J. W., II (1986) Thrombin:
Bioregulator Functions of Thrombin (Walz, D. A., Fenton, J. W., II,
and Shuman, M. A., eds.) in Ann. N. Y. Acad. Sci., Academy of
Sciences, New York, p. 5.
37.Aronson, D. L., Stevan, L., Ball, A. P., Franza,
B. R., Jr., and Finlayson, J. S. (1977) J. Clin. Invest.,
60, 1410-1418.
38.Drolet, D. W., Moon-McDermott, L., and Romig, T.
S. (1996) Nature Biotechnol., 14, 1021-1025.
39.Bruno, J. G., and Kiel, J. L. (1999) Biosens.
Bioelectron., 14, 457-464.
40.Jhaveri, S., Rajendran, M., and Ellington, A. D.
(2000) Nature Biotechnol., 18, 1293-1297.
41.Gebhardt, K., Shokraei, A., Babaie, E., and
Lindqvist, B. H. (2000) Biochemistry, 39, 7255-7265.
42.Iqbal, S. S., Mayo, M. W., Bruno, J. G., Bronk,
B. V., Batt, C. A., and Chambers, J. P. (2000) Biosens.
Bioelectron., 15, 549-578.