VPg Unlinkase, the Phosphodiesterase That Hydrolyzes the Bond between VPg and Picornavirus RNA: a Minimal Nucleic Moiety of the Substrate
A. Yu. Gulevich, R. A. Yusupova, and Yu. F. Drygin*
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119992 Russia; fax: (095) 939-3181; E-mail: drygin@belozersky.msu.ru* To whom correspondence should be addressed.
Received November 28, 2001; Revision received January 4, 2002
VPg unlinkase is an unusual eukaryotic enzyme that catalyzes hydrolysis of the phosphodiester bond between residues of the unique tyrosine of VPg (viral protein genome-linked) and the 5´-terminal uridylic acid of picornavirus RNA. Cellular targets of the VPg unlinking enzyme are yet unknown. To determine an essential nucleic part of the covalent linkage unit that is necessary for the VPg unlinkase reaction, the following derivatives of the encephalomyocarditis virus (EMCV) VPg-RNA complex were used: [125I]Kp-pUpUpGp, [125I]Kp-pUp, and [125I]Kp-pU (Kp is residual peptides bound to RNA after proteinase K treatment of VPg-RNA). [125I]K-peptides were unlinked from [125I]Kp-pUpUpGp and [125I]Kp-RNA with similar velocity, but [125I]Kp-pUp was split much slower. Under the same conditions [125I]Kp-pU was not dissociated at all. Thus, pUp is a minimal part of picornavirus RNA that is necessary for VPg unlinkase. We speculate that cellular substrates of the enzyme are phosphodiesters of oligo(poly)ribonucleotides and tyrosine or tyrosine peptides. In no case [125I]VPg-pU, [125I]VPg-pUp, and [125I]VPg-pUpUpGp were hydrolyzed by VPg unlinkase, in contrast with [125I]VPg-RNA and [125I]VPg-pUpUpGpApApApGp. We conclude that the whole VPg, when bound to trinucleotide (but not to heptanucleotide), protects the inter-polymeric phosphodiester bond against hydrolysis of the covalent linkage unit. We speculate that VPg unlinkase might repair covalent complexes of RNA and topoisomerases and trigger degradation process of the picornavirus RNA.
KEY WORDS: phosphodiesterase, picornavirus, VPg-RNA complex, radioactive iodination, substrate specificity
Abbreviations: EMCV) encephalomyocarditis virus; VPg) viral protein genome-linked; CLU) covalent linkage unit; Kp) residual peptides bound to RNA after proteinase K treatment of VPg-RNA; SE-HPLC) size exclusion high pressure liquid chromatography; SVE) snake venom (phosphodi)esterase.
The activity of an unusual HeLa cell phosphodiesterase that catalyzes
unlinking of VPg from poliomyelitis virus RNA (unlinking enzyme) was
discovered in 1978 [1]. This activity was found in
extracts of L cells, reticulocytes, and wheat germ, but not in E.
coli. The HeLa cell crude unlinking enzyme preparation, 280 times
enriched with the enzyme, catalyzed hydrolysis of the phosphodiester
bond between the unique VPg tyrosine residue and the 5´-terminal
uridylic acid of the poliovirus RNA [2, 3] faster than the inter-nucleotide bonds [1, 4]. However, VPg unlinkase
dissociated the covalent complex of VPg and the 5´-terminal
nonanucleotide of the poliovirus RNA markedly slower than the initial
VPg-RNA complex [1]. Moreover, the covalent linkage
unit (CLU) of VPg-pU, VPg-pUp, and Kp-pUp complexes was not cleaved by
the enzyme [4]. Later, VPg unlinking from
foot-and-mouth disease virus [5] and
encephalomyocarditis virus RNAs [6] was
demonstrated. VPg unlinkase of ascites Krebs II cells unlinked VPg and
K-peptides both from the EMC virus and poliovirus RNAs [6, 7].
Thus, an unusual phosphodiesterase of higher organisms (we did not succeed to uncover the VPg unlinkase activity of Saccharomyces cerevisiae) catalyzes specific hydrolysis of the phosphodiester bond between VPg and RNA of picornaviruses. It should be emphasized that the VPg unlinkase activity was revealed with the exogenous virus substrates, i.e., picornavirus RNA-protein covalent complexes. Cellular substrates that are structurally similar to the picornavirus substrates are still unknown.
We are studying the VPg unlinking enzyme from ascites carcinoma Krebs II cells [6-12]. We use the in vitro 125I-labeled Kp-RNA and VPg-RNA complexes of the encephalomyocarditis virus as substrates to purify and to characterize VPg unlinkase.
As expected, VPg unlinkase recognizes a nucleic constituent of the complex substrate ([1, 4, 7-10] and this paper). Yet, minimal magnitudes of the nucleic and protein moieties of the substrate have not been determined.
The goal of this work was to determine the minimal 5´-end fragment of RNA in the EMC virus Kp-RNA and VPg-RNA complexes that is necessary for the specific hydrolysis of the substrate by the enzyme under study. This subject is of considerable interest in the search for the cellular targets of VPg unlinkase, i.e., RNA-protein complexes that contain the covalent linkage unit identical to that of the picornavirus. In addition, knowledge of a minimal structure of the enzyme substrate might be useful for understanding of the VPg unlinkase function in the living cell and in picornavirus infection.
MATERIALS AND METHODS
Materials. RNase T1, RNase A, nuclease P1, snake venom (phosphodi)esterase (SVE), and agarose were from Sigma (USA); proteinase K from Boehringer Mannheim (Germany); acrylamide and 3-(4-hydroxyphenyl)propionic acid N-hydroxysuccinimide ester from Serva (Germany); N,N´-methylene-bis-acrylamide from Fluka (Switzerland); chloramine T and thin layer plates covered with Kieselgel 60 from Merck (Germany); Na125I from Izotop (Russia); and Hyperfilm-MP from Amersham (England).
VPg unlinkase substrates. EMC virus propagation and VPg-RNA isolation were performed as in [13]. Kp-RNA of EMC virus was produced from VPg-RNA by severe proteinase K treatment: 2 mg/ml of proteinase K in 10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 0.5% SDS for 2 h at 37°C.
Specific radioactive labeling of VPg or K-peptides bound to the EMC virus RNA was accomplished by the “two-steps-one-tube procedure” essentially as in [9]. Briefly, in the first stage of labeling, specific acylation of the aliphatic NH2 group in Kp(VPg)-RNA with excess of unlabeled 3-(4-hydroxyphenyl)propionic acid N-hydroxysuccinimide ester was achieved. Then 3-(4-hydroxyphenyl)propionic acid residues were labeled with 125I in the presence of chloramine T. The resulting radioactive substrates were examined by electrophoresis in 1% agarose gel.
Enzymatic reactions. The labeled Kp-RNA or VPg-RNA (0.01-0.1 µg) of EMC virus was completely hydrolyzed with 0.017 unit of nuclease P1 in 2 µl of water at 37°C for 1 h. To produce the free reference VPg or K-peptides, 2 µl of the P1 nuclease hydrolyzate was incubated with 8.5*10-5 units of SVE (1 µl) at 37°C for 1 h in 7 mM Tris-HCl, pH 7.5, and 1.7 mM MgCl2. To obtain Kp-pUp, 50 ng of Kp-RNA in 2 µl of water was treated with RNase A (1.25 mg/ml for 1 h at 20°C). Digestion of 10-100 ng of Kp-RNA or VPg-RNA with T1 RNase was carried out in 2 µl of water at 4°C for 1 h with 0.025 unit unless otherwise specified (see notes to figure legends).
Hydrolysis of substrates with VPg unlinkase was performed in TMM/200 (10 mM Tris-HCl, pH 7.5, 1.5 mM MgCl2, 5 mM 2-mercaptoethanol, 200 mM KCl) for 1 h at 37°C unless otherwise specified (activity of VPg unlinkase was practically the same in these buffer cocktails). Usually 4 µl of the VPg unlinkase preparation (SE-HPLC grade) [11] was added to 1-2 µl of the substrate.
Determination of enzymatic reaction products. Both thin layer chromatography and polyacrylamide gel electrophoresis were used to separate initial and final products of reactions. To analyze products of enzymatic reactions, usually 5-6 ng (~5000 cpm) of [125I]Kp-RNA or [125I]VPg-RNA after hydrolysis with enzymes were spotted onto TLC plates or ~50 ng (40,000 cpm) thereof were loaded on polyacrylamide gel. TLC was carried out on Kieselgel 60 plates (Merck, Germany) in butanol-1-water-acetic acid (3.75 : 1 : 1 v/v) system as described [8].
Electrophoresis was performed in polyacrylamide gel (15% acrylamide, 0.8% N,N´-methylene-bis-acrylamide, 7 M urea, 50 mM Tris-borate buffer, pH 8.3).
Amersham Hyperfilm-MP and intensifying yttrium screen (Mosmedfarmpreparat, Russia) were in use for autoradiography at -75°C for 12-24 h. Radioactive samples were counted in gamma-vials (Nuclear Chicago, USA).
RESULTS AND DISCUSSION
Assays of unlinking activity and the VPg unlinkase substrates. A complex and not easily obtained substrate makes assay of the VPg unlinkase activity very laborious. To monitor specific hydrolysis of the phosphodiester bond between VPg (Kp) and RNA, a separation and detection of the initial complex and products of its hydrolysis are ultimately required. There is no efficient universal method to satisfy these demands. Another approach is to track any constituent liberated (we choose the peptide part) from the complex. For this purpose we labeled specifically the protein moiety of the substrate with radioactive iodine and employed TLC on silica gel [8] and PAGE with 7 M urea.
Compared with the Bolton-Hunter reagent labeling of proteins [14], we succeeded to increase substantially (approximately two orders of magnitude) a selective radioactive iodination of Kp in the Kp-RNA complex,up to 1.5*106 dpm per µg (~0.4 pmol) of EMCV Kp-RNA by the “two-steps-one-tube procedure” [9]. To analyze unlinking activity of hundreds of fractions through the whole procedure of the VPg unlinkase isolation, we used labeled Kp-RNA, instead of the more complicated VPg-RNA substrate, and very efficient TLC on silica gel. After specific acylation of the N-terminal aliphatic NH2 group in Kp(VPg)-RNA by excess of unlabeled 3-(4-hydroxyphenyl)propionic acid N-hydroxysuccinimide ester and following labeling with 125I, the amino acylated free K-peptides became more hydrophobic and were well separated by silica gel TLC from each other (Fig. 1, lanes 3 and 4) and from K-peptides linked with the 5´-end uridine phosphate (or the 5´-end phosphate and uridine-5´,3´-diphosphate, data not shown). At the same time Kp-RNA, as well as Kp-oligonucleotides, do not move in this chromatography system and are retained at the start.
Unlinking enzyme was isolated from mouse ascites Krebs II cells by a method including ammonium sulfate fractionation, pH-fractionation, cation exchange chromatography on CM-52 cellulose, chromatofocusing, and size exclusion HPLC [11].Fig. 1. Selectivity of K-peptide radioactive iodination in the complex substrate Kp-RNA and unlinking of K-peptides by the purified VPg-unlinkase. EMCV [125I]Kp-RNA (6.25 ng, ~5000 cpm, 1 µl) was incubated at 37°C for 1 h with water (control) or hydrolytic enzymes. Then reaction cocktails (5 µl) were spotted onto a thin layer silica gel plate. Chromatography was continued until the front of the liquid system (see “Materials and Methods”) reached the 9 cm mark. Lanes: 1) after addition of water (4 µl); 2) nuclease P1 was added to hydrolyze RNA completely; 3) treatment with nuclease P1 was followed by SVE to produce free K-peptides; 4) SE-HPLC grade VPg unlinkase was added to the substrate; 5) nuclease P1 treatment was followed by SVE as in 3 and then released K-peptides were digested by proteinase K (see “Materials and Methods”); 6) [125I]Kp-RNA treated repeatedly by proteinase K alone as in 5. Notice, EMC virus RNA protects K-peptides against profound digestion by proteinase K (compare lanes 1, 3, 5, and 6).
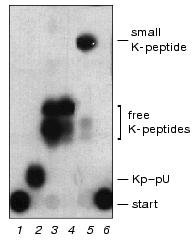
It is well documented that the unlinked free VPg is rapidly hydrolyzed by some unidentified cell protease [4, 8, 11, 15] in crude VPg unlinkase preparations. However, in the covalent complex the EMC virus RNA protects VPg (and residual K-peptides) against proteolysis. This explains heterogeneity of K-peptides produced after complete digestion of VPg, while bound to the virus RNA, by proteinase K (Fig. 1, see also Fig. 3). K-peptide heterogeneity is particularly obvious when the highly radioactive substrate and powerful separation methods (TLC on Kieselgel plates and PAGE) are employed. Interestingly, even the 5´ terminal uridylic acid linked with K-peptides blocks complete Kp (VPg) hydrolysis by proteinase K, whereas free K-peptides become sensitive to the repeated treatment by this protease (Fig. 1, compare lanes 3 and 5). This fact has to be kept in mind through a structural work with proteins modified by nucleotides.
Earlier we showed that free VPg of encephalomyocarditis virus is positively charged in a neutral medium [13]. Figure 2 shows that after acylation of its amino groups by 2,6-diiodo-1-hydroxyphenyl-4-propionate, unlinked VPg acquires a small negative charge and moves slowly in a polyacrylamide gel toward the anode. Addition of proteases inhibitors (a mixture of aprotinin, pepstatin, leupeptin, soybean trypsin inhibitor, 10 µg/ml each) to the VPg unlinkase assay cocktail did not change the pattern of products (data not shown). Mobility of VPg released from the VPg-RNA complex by the enzyme under study and after P1 + SVE treatment are similar (see Figs. 2 and 5). We conclude that the purified VPg unlinkase preparation [11] lacks proteolytic activity.
The roughly estimated Km of VPg-RNA was about 0.1 µM. Free VPg appeared at the very beginning of the specific hydrolysis in the assay cocktail. To economize on the not easily attainable substrate, its concentration in the assay was usually substantially lower than Km.Fig. 2. Kinetics of VPg unlinking from EMC virus [125I]VPg-RNA by the purified VPg-unlinkase (examined by 7 M urea/15% PAGE). EMC virus [125I]VPg-RNA (1 µg, ~10,000 cpm, 1 µl of water solution; a preparation with low specific radioactivity was used for kinetic considerations) was incubated at 37°C for 1 h with VPg unlinkase or in 0.5× TMM/200 buffer. Lanes: 1) control, 0.5× .MM/200 buffer; 2, 3) nuclease P1 alone and P1+ SVE, respectively (as in Fig. 1, lanes 2 and 3); 4-7) unlinking of VPg by VPg unlinkase for 30, 20, 15, and 10 min, respectively; 8) T1 RNase (0.025 unit) was added to [125I]VPg-RNA and mixture was incubated in 2 µl of water at 4°C for 1 h to obtain reference VPg-oligonucleotides. SE-HPLC grade VPg unlinkase preparation has no proteolytic activity (see Fig. 5 also), but a trace amount of purine specific nuclease can be detected.
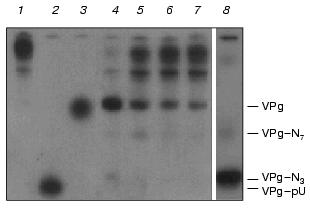
Studying the VPg unlinkase reaction kinetics, at the high substrate concentrations we found by PAGE that the final enzyme preparation is admixed with a trace of guanine specific nuclease (Fig. 2). However, this admixed nucleolytic activity generated mainly the 5´ terminal long fragments of the EMC virus VPg-RNA and negligible amount of fragments that matched VPg-pUpUpGp and VPg-pUpUpGpApApApGp produced by T1 RNase. The same results were obtained with Kp-RNA as substrate.
Actually, this admixture did not interfere with VPg unlinkase. In addition, Kp(VPg)-pU and/or Kp(VPg)-pUp were never detected as by-products of the substrate incubation with VPg unlinkase. We found also that the 5´ terminal fragments as long as VPg-pUpUpGpApApApGp and longer were readily and specifically hydrolyzed by the examined phosphodiesterase.
The minimal nucleic part of the substrate. A minimal structure of the covalent linkage unit of the VPg-RNA complex that suffices for the VPg unlinkase substrate is unknown. Taking into consideration that the enzyme recognizes rather lengthy RNA [1, 8, 10, 12], one can conclude, that step-by-step shortening of the 5´ terminal RNA part of the substrate will suppress the VPg unlinking reaction. To find a minimal size of the nucleic constituent that is necessary for the unlinking reaction, we first dissected the EMC virus Kp-RNA by either T1 RNase or nuclease P1 and RNase A, then the products were treated with VPg unlinkase.
As follows from Fig. 3, free K-peptides and those bound to the 5´ end (oligo)nucleotides were well separated from each other by 7 M urea/15% PAGE. Notice that K-peptides linked with pU move slightly faster than free K-peptides due to the larger negative charge. However, Kp-pUpUpGp moves under the same conditions slower than Kp-pU and Kp-pUp because of larger size. In addition, lengthening of the nucleic part results in the lower resolution of oligonucleotide-peptides with K-peptides of different size. Therefore, free K-peptides are represented by four bands, compounds Kp-pU and Kp-pUp by three bands, and Kp-pUpUpGp with one broad band.
The pattern of the 5´ end Kp-oligonucleotides produced by RNase T1 under restricted conditions showed a good matching with the pattern predicted from the primary and secondary structure of EMCV RNA [16] (Figs. 2-4). As follows from the primary structure of EMCV RNA, the first guanine residue is at the third position. Hence, the products of the complete digestion of Kp-RNA by RNase T1, RNase A, and nuclease P1 have to correspond to Kp-pUpUpGp, Kp-pUp and Kp-pU, respectively. Identity of Kp-pUpUpGp was initially verified its finding as the only product after complete digestion of Kp-RNA by RNase T1, by its compatible mobility in PAGE with mobility of Kp-pU and Kp-pUp (see Figs. 3 and 4) and finally, by its structural analysis [12].Fig. 3. VPg unlinkase readily hydrolyzes Kp-pUpUpGp, but not Kp-pU. EMC virus [125I]Kp-RNA (50 ng, ~40,000 cpm) was incubated with TMM/200 (4 µl) for 1 h at 37°C (control, 1) or with nuclease P1 (see “Materials and Methods”) (2), or nuclease P1 followed by SVE (3); RNase T1 (1 µl, 0.025 unit) for 1 h at 20°C (4); VPg unlinkase preparation for 30 min at 37°C (5); nuclease P1 followed by VPg unlinkase for 1 h at 37°C (6). It should be emphasized that no free K-peptides were found; RNase T1 (as in 4) followed by VPg unlinkase (as in 6) (7). Notice, K-peptides bound to tri- and longer oligonucleotides are readily hydrolyzed by VPg unlinkase. Samples were examined by 7 M urea/15% PAGE.
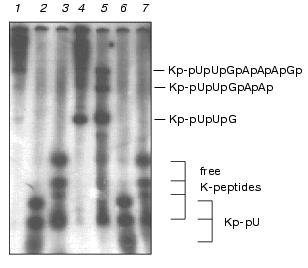
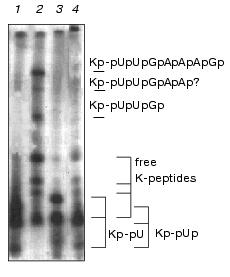
Ascites unlinking enzyme digested Kp-pUpUpGp at virtually the same efficiency as EMCV Kp-RNA (Fig. 3). Comparing results of lanes 3 and 4 (Fig. 4) shows that Kp-pU is not a substrate for VPg unlinkase, but Kp-pUp is slowly hydrolyzed by an unlinking enzyme.Fig. 4. A minimal nucleic part of the substrate for VPg unlinkase is Kp-pUp. EMC virus [125I]Kp-RNA (50 ng, ~40,000 cpm, 1 µl, in water) was incubated with: RNase A to produce Kp-pUp (1); VPg unlinkase preparation (2); nuclease P1 followed by VPg unlinkase (3); RNase A (as in 1) followed by VPg unlinkase (4). All samples were examined using 7 M urea/15% PAGE.
This finding is in contradiction with earlier published data for the HeLa cell unlinking enzyme [1]. We explain this discrepancy by much higher sensitivity of our analysis because the specific radioactivity of the substrate we used was more than 1000-fold higher.
It should be stressed that we have never seen digestion of Kp-pU (Figs. 3 and 4) by the enzyme, whereas Kp-RNA and Kp-pUpUpGp were hydrolyzed quantitatively under the same conditions. We conclude that pUp is the minimal part of the nucleic constituent of the EMC virus Kp-RNA that is necessary for the enzyme reaction. But whether VPg unlinkase recognizes precisely pUp in the CLU or perhaps another nucleotide, such as pC, pA, or pG, is not known.
Suggested structure for the VPg unlinkase cellular substrates. Earlier we showed that synthetic phosphodiesters formed by pU and tyrosine with protected amino (Bz, Ac) and carboxy (Et, Met) groups as well as by pTCTCTCCTCTTCCCTCC and protected tyrosine are not hydrolyzed by VPg unlinkase [9]. Moreover, phosphodiesters of pT13C2 either with protected tyrosine or with p-chlorophenol and methanol, as also had pT13C2 alone, had little or no effect [17] on the unlinking reaction when these substances were added to the assay cocktail in the 10-fold molar excess over the substrate.
Hence, an unlinking enzyme recognizes ribonucleic derivatives of tyrosine (or tyrosine peptides); deoxyribonucleic phosphodiesters of tyrosine are not hydrolyzed by the enzyme and do not compete with the substrate.
Finally, the unlinking enzyme hydrolyzes the phosphodiester bond between picornavirus VPg and RNA, but not in the comovirus VPg-RNA complex [7], where VPg is bound to the 5´ terminal uridine phosphate residue of RNA via a serine residue [18, 19]. Whether another related cellular enzyme exists that unlinks comovirus VPg from RNA is not yet known.
As mentioned earlier, integrity of VPg in the complex substrate is dispensable for VPg unlinkase. It seems that all K-peptides produced from VPg in the complex with RNA are equally unlinked from it. However, whether only the tyrosine residue of VPg, when bound to RNA is sufficient for the unlinking enzyme substrate or either di- or tripeptide is necessary remains obscure. Presumably, synthetic phosphodiesters of tyrosine and (oligo)ribonucleotides could help to clarify this. Of course, these substrates could simplify study of VPg unlinkase. This work is in progress.
Thus, the phosphodiesterase under examination reveals a unique specificity to the structure of the covalent linkage unit of the substrate. Summing up data on the unlinking enzyme specificity, one might speculate that the enzyme is a member of a particular group of eukaryotic phosphodiesterases that are involved in metabolism of some cellular phosphodiesters of polyribonucleotides and peptides.
Covalent VPg-RNA complexes are peculiar to many groups of human, animal, and plant viruses. Their formation-dissociation mechanisms appeared to be general ones inside a class [17]. It is attractive to speculate that these mechanisms are involved in dissociation of yet unknown cellular covalent complexes of RNA and protein. Knowledge of the substrate specificity of the enzymes involved (unlinking enzymes?!) might be of help in searching for their cellular targets. However, VPg unlinkase does not catalyze hydrolysis of phosphodiesters formed by mononucleotides and VPg or the tyrosine peptides. Thus, there is a low probability that this enzyme is involved in a regulation activity of uridylated proteins similar to bacterial glutamine synthetase [20].
The idea that VPg unlinkase is involved in the cleavage of dead intermediate covalent complexes of some RNA with poisoned cell DNA-topoisomerases is not without appeal. Complexes between RNA and DNA-topoisomerase III [21] or DNA-topoisomerases IIalpha and IIbeta [22] can be obtained in vitro. There is a high probability that topoisomerases in the intermediate complexes are bound to RNA via a tyrosine residue by a phosphodiester bond, similar to the active intermediate phosphodiesters of topoisomerases and DNA ([23], see also [17] and references therein), because, while alive, they are able to catalyze trans-esterification reaction thus restoring inter-nucleotide bond [21]. Recently similar eukaryotic phosphodiesterase that catalyzes specific hydrolysis of CLU in the dead-end complex formed by TopoI and DNA was isolated from eukaryotes [24]. It was proposed that this enzyme is involved in the restoration process of an inter-nucleotide bond by dissociating the dead topoisomerase from the covalent complex with DNA.
It must be emphasized that the VPg unlinkase activity was discovered with an exogenous substrate, i.e., picornavirus VPg-RNA covalent complex. To determine a role of this enzyme in the living cell and in the picornavirus infection, identification of the coding gene and its mutagenesis are necessary.
Possible roles of VPg unlinkase in picornavirus infection. It remains unknown why the picornavirus polysomal RNA is deprived of VPg. Thus, several functions of the unlinking enzyme during picornavirus infection have been suggested since its discovery, including triggering of the picornavirus RNA translation, negative regulation of the viral RNA encapsidation, or replication [1, 4, 5, 8, 15]. VPg unlinkase might be another member in a set of cellular factors [25] implicated in virus outgrowth as well. It is known that activity of an unlinking enzyme is associated with a membrane fraction [1, 4, 6, 8]. Nothing is known, however, concerning whether an unlinking enzyme is modified after infection. Our preliminary data indicate that levels of the unlinking activity in infected and non-infected cells are closely related. Ultimately, a role for VPg unlinkase in picornavirus infection development is obscure.
Regarding the data obtained in this work, we confirmed that the ascites VPg unlinkase does not cleave VPg-pU or VPg-pUp produced from encephalomyocarditis VPg-RNA, as was earlier found for the HeLa unlinking enzyme and for the same compounds of the polio virus [1, 2]. Moreover, we have discovered that VPg-pUpUpGp is resistant to VPg unlinkase, in contrast to Kp-pUpUpGp, which is readily cleaved by the enzyme (Fig. 5, see also Fig. 2, lane 4).
Paradoxically, the extended 5´-end VPg-oligonucleotide fragments are good substrates of VPg unlinkase in vitro [1, 7]. As pointed out above, the ascites VPg unlinkase did specifically hydrolyze the EMC virus VPg-pUpUpGpApApApGp (Fig. 2) and the HeLa unlinking enzyme cleaved the polio virus VPg-pUpUpApApApApCpApGp. Hence, a spatial configuration of the covalent linkage unit in the extended substrate (VPg-pUpUpGpApApApGp) itself or in the complex with VPg unlinkase have to differ from that in the short derivative VPg-pUpUpGp. This stereo-reorganization may be induced by the enzyme on binding with CLU in the VPg-oligonucleotide substrates either by interaction of proximal (or distal?) pApA, pApG sequences with VPg, thus neutralizing protection of CLU by VPg, or with the 5´-end pUpU.Fig. 5. VPg unlinkase does not hydrolyze VPg-pUpUpGp and VPg-pU. EMCV [125I]VPg-RNA (70 ng, ~10,000 cpm, 1 µl) was incubated with: 4 µl of TMM/200 for 1 h at 37°C (control) (1); nuclease P1 (2); nuclease P1 followed by SVE (3); 1 µl of RNase T1 (0.025 unit) for 1 h at 20°C (4); nuclease P1 followed by VPg unlinkase (5); RNase T1 (as in 4) followed by VPg unlinkase (6); VPg unlinkase (7). Reaction cocktails (5 µl) were spotted onto thin layer silica gel plate and chromatography was continued. Notice, in contrast to Kp-derivatives, VPg-pUpUpGp is resistant to the unlinking enzyme (compare with Fig. 3).
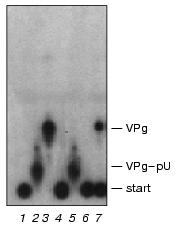
Considering the data described above and that the pUpU sequence is at the 5´ end RNA of all picornaviruses (Genbank, NCBI), one can explain why VPg-pUpU is found as a major product during the initiation of the picornavirus replication in vivo [26] or in crude membrane fraction [27-29]. VPg-pUpU has to be resistant to the cleavage by VPg unlinkase like VPg-pUpUpGp [1, 4, 6, 8]. In this connection it seems doubtful whether VPg unlinkase acts as a negative regulator of the picornavirus RNA initiation of replication.
Finally, VPg unlinking resembles decapping of cellular mRNAs. Recent findings proved that decapping of cellular messages triggers their decay in vivo and in cell extracts in both 3´-->5´ and 5´-->3´ directions [30, 31]. In this case VPg unlinkase, one can propose, might be a part of the anti virus cellular defense system. Mechanisms of a cell message decay consist of consecutive partial deadenylylation, decapping, endo- and exonucleolytic steps with 5´-->3´ exonuclease or 3´-->5´ multiprotein exosome [30, 32]. Yet, VPg unlinking does not depend on deadenylation in vitro [9], as it takes place for many cellular mRNAs. However, one can note that a scheme where virus mRNA degradation was not preceded by shortening of the poly(A) tail, and where the 5´-end sequences degraded more rapidly than those of the 3´-end, was described for the simplex virus infected cells [33].
The authors thank Drs. A. Bogdanov, V. Agol and A. Antonov for critical comments on the manuscript and S. Bubenschikova for technical assistance with HPLC.
This work was supported in part by the Russian Foundation for Basic Research (grant 01-04-48652a).
REFERENCES
1.Ambros, V., Petterson, R. F., and Baltimore, D.
(1978) Cell, 15, 1439-1446.
2.Lee, Y. F., Nomoto, A., and Wimmer, E. (1976)
Progr. Nucleic Acids Res. Mol. Biol., 19, 89-96.
3.Flanegan, J. B., Petterson, R. F., Ambros, V.,
Hewlett, M. G., and Baltimore, D. (1977) Proc. Natl. Acad. Sci.
USA, 74, 961-965.
4.Ambros, V., and Baltimore, D. (1980) J. Biol.
Chem., 255, 6739-6744.
5.Sangar, D. V., Bryant, J., Harris, T. J., Brown,
F., and Rowlands, D. J. (1981) J. Virol., 39, 67-74.
6.Drygin, Yu. F., and Siyanova, E. Yu. (1986)
Biokhimiya, 51, 249-258.
7.Drygin, Yu. F., Shabanov, A., and Bogdanov, A.
(1988) FEBS Lett., 239, 343-346.
8.Drygin, Yu. F., Shabanov, A. A., and Bogdanov, A.
A. (1988) Biokhimiya, 53, 1371-1379.
9.Shabanov, A. A., Kalyuzhny, A. A., Gottikh, M. B.,
and Drygin, Yu. F. (1996) Biochemistry (Moscow), 61,
794-803.
10.Viryasov, M. B., Yusupova, R. A., Shatskaya, G.
S., and Drygin, Yu. F. (1997) Chromatographia, 45,
133-137.
11.Yusupova, R. A., Gulevich, A. Yu., and Drygin,
Yu. F. (2000) Biochemistry (Moscow), 65, 1219-1226.
12.Gulevich, A. Yu., Yusupova, R. A., and Drygin,
Yu. F. (2001) Biochemistry (Moscow), 66, 345-349.
13.Drygin, Yu. F., Vartapetian, A. B., and Chumakov,
K. M. (1979) Mol. Biol. (Moscow), 13, 777-787.
14.Bolton, A. E., and Hunter, W. M. (1973)
Biochem. J., 133, 529-539.
15.Dorner, A. J., Rothberg, P. G., and Wimmer, E.
(1981) FEBS Lett., 132, 219-223.
16.Vartapetian, A. B., Mankin, A. S., Skripkin, E.
A., Chumakov, K. M., Smirnov, V. D., and Bogdanov, A. A. (1983)
Gene, 26, 189-195.
17.Drygin, Yu. F. (1998) Nucleic Acids
Res., 26, 4791-4796.
18.Drygin, Yu. F., Sapotsky, M. V., and Bogdanov, A.
A. (1987) FEBS Lett., 215, 247-251.
19.Jaegle, M., Wellink, J., and Goldbach, R. (1987)
J. Gen. Virol., 68, 627-632.
20.Adler, P. A., Purich, D., and Stadman, E. R.
(1975) J. Biol. Chem., 250, 6264-6272.
21.Wang, H., Di Gate, R. J., and Seeman, N. C.
(1996) Proc. Natl. Acad. Sci. USA, 93, 9477-9482.
22.Wang, Y., Knudsen, B. R., Bjergbaek, L.,
Westergaard, O., and Andersen, A. H. (1999) J. Biol. Chem.,
274, 22839-22846.
23.Tse, Y.-C., Kirkegaard, K., and Wang, J. C.
(1980) J. Biol. Chem., 255, 5560-5565.
24.Yang, S.-W., Burgin, A. B., Jr., Huizenga, B. N.,
Robertson, C. A., Yao, K. C., and Nash, H. A. (1996) Proc. Natl.
Acad. Sci. USA, 93, 11534-11539.
25.Andino, R., Boddeker, N., Silvera, D., and
Gamarnik, A. V. (1999) Trends Microbiol., 7, 76-82.
26.Crawford, N. M., and Baltimore, D. (1983)
Proc. Natl. Acad. Sci. USA, 80, 7452-7455.
27.Takegami, T., Kuhn, R. J., Anderson, C. W., and
Wimmer, E. (1983) Proc. Natl. Acad. Sci. USA, 80,
7447-7451.
28.Vartapetian, A. B., Koonin, E. V., Agol, V. I.,
and Bogdanov, A. A. (1984) EMBO J., 3, 2583-2588.
29.Takeda, N., Kuhn, R. J., Yang, C.-F., Takegami,
T., and Wimmer, E. (1986) J. Virol., 60, 43-53.
30.Decker, C. J., and Parker, R. (1994) Trends
Biochem., 19, 336-340.
31.Carpousis, A. J., Vanzo, N. F., and Raynal, L. C.
(1999) Trends Genet., 15, 24-28.
32.Allmang, C., Petfalski, E., Podtelejnikov, A.,
Mann, M., Tollervey, D., and Mitchell, P. (1999) Gen. Dev.,
13, 2148-2158.
33.Karr, B. M., and Read, G. S. (1999)
Virology, 64, 195-204.