Interaction of Influenza A Virus M1 Matrix Protein with Caspases
O. P. Zhirnov1*, A. L. Ksenofontov1, S. G. Kuzmina1, and H. D. Klenk2
1Ivanovski Virology Research Institute, Russian Academy of Medical Sciences, ul. Gamalei 16, Moscow, 123098 Russia; fax: (095) 190-3058; E-mail: olegzhirnov@hotmail.com2Robert-Koch-Institüt, 13353 Berlin, Institüt für Virologie, Philipps-Universität, 35037 Marburg, Germany; fax: (49) 6421-2868962; E-mail: klenk@mailer.uni-marburg.de
* To whom correspondence should be addressed.
Received October 3, 2001; Revision received February 1, 2002
In this investigation, an ability of influenza A virus M1 matrix protein to bind intracellular caspases, the key enzymes of cell apoptosis, has been examined. Protein-protein binding on polystyrene plates and polyvinyl pyrrolidone membrane was employed for this purpose. Under a comparative study of caspases-3, -6, -7, -8 influenza virus M1 protein specifically bound caspase-8 and weakly bound caspase-7. Using a computer analysis of the N-terminal region of M1 protein, a site similar to the anti-caspase site of baculovirus p35 protein, which inhibits caspases and displays antiapoptotic activity, was identified. These results are in good agreement with the supposition that influenza virus M1 protein is involved in a caspase-8-mediated apoptosis pathway in influenza virus infected cells.
KEY WORDS: influenza virus, apoptosis, matrix protein, caspases
Influenza virus belongs to a lipid-containing capsid virus family. It is comprised of four main structural proteins: nucleocapsid protein (NP), M1 matrix protein, surface glycoproteins HA and NA, and five minor components: three polymerase polypeptides (PB1, PB2, PA), ion channel protein (M2), and nuclear export protein (NEP, NS2). Virus particles contain 500 (glycoproteins) to 1000-3000 (M1 and NP, respectively) molecules of main structural proteins, and 20-40 molecules of minor polypeptides for one virion [1].
M1 protein forms a virion protein matrix that is located under the lipid membrane [2, 3]. M1 matrix plays a mediator role between the lipid capsule and viral ribonucleoprotein (RNP). To fulfill the integrating function, M1 protein contains an excess of positively charged arginine residues, by means of which M1 interacts with viral RNA and negatively charged phospholipid knob in the membrane, and the hydrophobic regions responsible for additional interaction with virion lipid membrane [4, 5].
Besides the structural role in the virus, M1 protein has a number of regulatory functions in the infected cells. It participates in the release and activation of transcription of viral RNP when the virus penetrates into the target cell [6], regulates the migration of viral RNP from the nucleus and its subsequent cytoplasmic transport to the virion assembling sites [7, 8], and takes part in the regulation of viral particle assembly [9]. In accordance with the polyfunctional nature of M1 matrix protein, a number of structure-functioning domains have been identified in its sequence: RNA-binding [10], histone-binding [11], protease-binding [5, 12], “zinc finger” sequence [13], signal of intranuclear transport and nuclear accumulation [14, 15].
It has been recently determined that influenza virus causes apoptosis of the infected cells at the latest stages of infection [16, 17]. It was also shown that influenza infection triggers a caspase-8 induced apoptosis pathway [18]. However, the cell apoptosis initiation mechanism and the role of viral proteins in this process still remain unclear. There is some evidence that non-structural viral protein NS1 (25 kD) is able to initiate apoptosis under its expression in the cells alone [19], but this protein, on the contrary, displays an anti-protease activity in infected cells [20]. It can be assumed that influenza virus synthesizes protein(s) preventing cell apoptosis at early infection stages so as to perform its replication before apoptosis is triggered.
One possible candidate to be considered for the role of apoptosis factor (it is still unknown whether inducer or suppressor) is M1 matrix protein, which impaired a caspase-8 mediated apoptosis, induced by tissue necrosis factor TNF-alpha under expression in human carcinoma cells Hep-2, as we mentioned previously [21]. In respect to this and also taking into account that M1 protein displays a protease-binding activity, we have investigated the affinity of M1 influenza protein towards various cellular caspases. Under a comparative study of caspases-3, -6, -7, and -8 we have discovered that M1 selectively interacted with caspase-8. This observation is consistent with the fact that apoptosis in influenza infected cells is activated by a caspase-8-dependent pathway, with a possible participation of M1 viral protein.
MATERIALS AND METHODS
Viruses and cells. Human influenza A viruses A/FPV/34 (H7N7), A/Aichi/68 (H3N2), B/Hong Kong/72, and C/USSR/77 passaged in chicken embryos as described earlier [22] were used in this work. Viruses were purified for polypeptide analysis by differential precipitation using 20% sucrose solution as was previously reported [22].
Isolation of M1 influenza viral protein. M1 influenza viral protein was isolated from virus species produced in the chicken embryos by acidic solubilization technique as was described in an earlier paper [23]. Purity of the resulting M1 preparation was estimated by polyacrylamide gel electrophoresis.
Protein electrophoresis in polyacrylamide gel. Polyacrylamide gel electrophoresis (PAGE) was performed in a polyacrylamide gel containing SDS using a miniaturized setup (7 × 8 cm) as described previously [10]. Prior to the electrophoresis, the analyzed samples were dissolved in buffer containing 1% SDS, 10 mM dithiothreitol (DTT), and 10% glycerol.
Study of the interaction of M1 protein with caspases on a solid phase. Polystyrene microtiter plates (HighTech, USA) were used for the investigation of M1-caspase interaction. Isolated M1 protein was adsorbed in the microwells during the overnight incubation (50 µl per well) of 1 µg/ml M1 protein solution; after that the microwell surface was saturated with a 5% bovine serum albumin (BSA) solution applied for 2 h. Solutions (50 µl) of purified caspases-3, -6, -7, and -8 taken at various dilutions (2.1-0.06 µg/ml) in phosphate buffer containing 0.1% nonionic detergent Tween-20 and 0.2% BSA was then added in the microwells and incubated for 2 h at 10°C. The aliquots of purified caspases were generously provided by Prof. G. S. Salvesen (The Burnham Institute, CA, USA). Microtiter plates were further rinsed with phosphate buffer containing 10 mM Na2HPO4/NaH2PO4, pH 7.2, 2.7 mM KCl, 137 mM NaCl, and 0.1% Tween-20 and incubated with anti-caspase-3 (polyclonal rabbit IgG; Santa Cruz, USA), anti-caspase-8 (goat IgG towards peptides C-20 and S-19; Santa Cruz), anti-caspase-6 (mouse monoclonal IgG, clone B93-4; Pharmingen, USA), and anti-caspase-7 (mouse monoclonal IgG, clone B94-1; Pharmingen) antibodies. The formation of immune complexes was detected spectrophotometrically by a reaction of secondary antibodies conjugated to horseradish peroxidase (HRP, Dako, Belgium), and using tetramethylbenzidine as the substrate (Sigma, USA). Extent of the reaction was assessed by a color intensity, registered by a microreader (model 550, BioRad, USA) at 450 nm.
Interaction of M1 protein with caspase-8 on nitrocellulose membrane. Purified samples of A, B, and C viruses and matrix protein M1 were fractionated using PAGE and then transferred onto polyvinylidene difluoride (PVDF) membrane by semi-dried method as described previously [24]. The membrane was then incubated with a specific ligand caspase-8 (Pharmingen, 0.8 µg/ml) in phosphate buffer containing 0.5% BSA, 10 mM Tris-HCl, pH 7.5, for 12 h at 10°C. After that the membrane was carefully rinsed with phosphate buffer. Caspase-8 adsorbed on the membrane was identified using goat anti-caspase-8 antibodies and a conjugate of secondary anti-goat antibodies with HRP (Dako) by the enhanced chemiluminescence (ECL) method with ECL-supersubstrate (Pierce, USA).
Computer search for the homologous supermotifs. A comparison of the primary structures was performed by a multiple sequence alignment program (www.genebee.msu.su/services/malign reduced.html) using a pairwise comparison [26] with a Niedelman-Wunsch algorithm and amino acid sequence matrix [27]. The similarity of the aligned amino acid pair was defined by a coefficient depicting their contribution into the tertiary structure of a selected homological region (supermotif). Considering the increase of similarity between the analyzed amino acids, the coefficient could fall into one of three ranges: 1SD below the average value of Johnson's matrix (*), or 1SD (+) and 2SD (*) above that one, respectively (see Fig. 3) [26].
RESULTS
In the first part of the present investigation, the ability of influenza A virus M1 matrix protein to bind different caspases was investigated. For this purpose, M1 protein was isolated from viral particles using a selective solubilization at acidic pH. As we have shown previously, M1 isolated according to this technique did not aggregate and maintained its inhibitory effect towards polymerase activity of viral ribonucleoprotein (RNP), despite the high hydrophobicity of the former [23]. Purity estimation of M1 preparation using PAGE revealed that the isolated M1 was of high purity (~95%) [23]. Then the obtained soluble M1 polypeptide was used as a specific target for caspases-3, -6, -7, and -8. Two aspects should be mentioned. First, M1 was not exposed to a denaturing treatment for the preservation of its native conformation when adsorbed on the microtiter plate. Second, the active form of the caspases involved in our experiments was constituted by two proteolytic subunits, and, in addition, caspase-8 did not contain procaspase domain DED (Death Effector Domain) [25]. After the reaction and careful rinsing of the wells, the presence of caspases (which in this case indicated their binding with M1) was detected immunologically using caspase-specific antibodies.
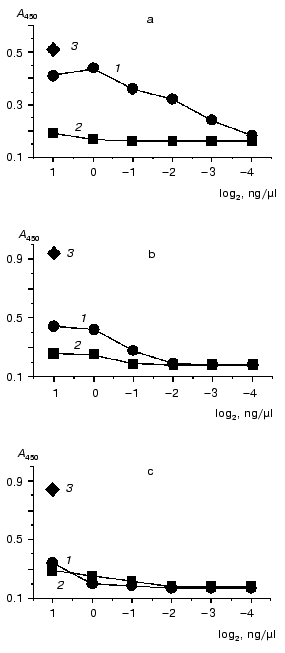
As evidenced by the results (Fig. 1), a detectable positive signal was observed in the case of caspase-8 and was absent under the incubation of M1 protein with caspases-3 and -6. A minor signal (exceeding the negative control signal to some extent, but significantly lower than the positive control signal) was observed for caspase-7, and this was considered as a weak nonspecific interaction of M1 and caspase-7 under the experiment conditions. The data shown in Fig. 1 were obtained with M1 from avian influenza virus A/FPV/34 (H7N7). Similar observations, however, were also obtained for M1 isolated from human influenza virus A/Aichi/68 (H3N2) (data not shown). The results indicated that influenza A virus M1 matrix protein displays an appreciable affinity towards caspase-8 and is capable of forming stable complexes with the latter.
In the second part of the investigation comparative experiments with influenza viruses A, B, and C were carried out for confirmation of specific interaction between influenza A virus M1 protein and caspase-8. For this purpose, Western blot analysis technique and a method of protein entrapment were applied, by means of which an ability of influenza virus structural proteins of the above mentioned types to bind caspase-8 was analyzed. Influenza virus preparations were dissociated using the ionic detergent SDS, then subjected to PAGE, and transferred onto the PVDF membrane, which was incubated with caspase-8. The latter, adsorbed on specific sites of the membrane was identified using the antibodies towards caspase-8. The results obtained are presented in Fig. 2. First, evaluated A, B, and C viruses had a number of main structural polypeptides typical for influenza viruses: NP (56-62 kD), HA1 (50 kD), HA2, and M1 (27 kD), electrophoretic mobility of which was characterized by a virus-specific variability (Fig. 2a). Second, a preparation of caspase-8 essentially consisted of its activated form constituted by two subunits p34 and p20, and contained only a negligible amount of uncleaved form p60 (Fig. 2b, lane K). Third, caspase-8 was selectively adsorbed by influenza A virus M1 matrix protein and did not interact with other structural viral proteins, at least with such main structural polypeptides as HA1, HA2, and NP. Fourth, M1 matrix protein of B/Hong Kong/72 and C/USSR/77 influenza viruses did not display an ability to bind caspase-8, contrary to that demonstrated by influenza A virus (Fig. 2b, lanes B and C). Thus, these observations confirmed the conclusion on the specific binding of caspase-8 by influenza A virus M1 matrix protein, and indicated the lack of this feature of the corresponding M1 matrix proteins of influenza B and C viruses.Fig. 1. A comparative binding of caspases-3, -6, -7, and -8 by influenza A virus M1 matrix protein. M1 matrix protein from A/FPV/34 (1) virus or BSA (control, 2) were adsorbed on the microtiter plate as a support, which was then incubated with various concentration (abscissa axis) of caspase-8 (a), -7 (b), -3 and -6 (c). 3) Signal given by a positive control, obtained when a corresponding caspase at the concentration of 2 ng/µl was used as a support. The adsorbed caspase was detected immunologically by using the HRP conjugate of secondary antibodies, colorimetric detection, and tetramethylbenzidine as a substrate. Reaction intensiveness (ordinate axis) was defined by the variation of absorbance at 450 nm. Since both caspase-3 and -6 had an almost identical value indicating the lack of binding with M1, an averaged curve represents these two caspases.
With respect to the discovery of caspase-8-binding properties of influenza A virus M1 matrix protein, we carried out a computer search for structural analogies of viral M1 with other known apoptosis inhibitors. Special attention was given to the antiapoptotic proteins from the caspase inhibitors group. The preliminary analysis (Fig. 3) revealed a structural similarity of the N-terminal region of M1 (amino acid residues 20-32) with a region of Autographa californica baculovirus p35 protein, displaying the caspase inhibitory properties, also including caspase-8 [32], and playing a role of viral apoptosis inhibitor [33]. The revealed homology region was located within residues 63-75 of the p35 sequence, thus being adjacent to the region (amino acid residues 84-88) involved in the formation of active anti-caspase center [34]. The similarity between p35 and M1 in the region of the supermotif was characterized with a coefficient of power of 4.45, and homologous amino acids were found in 43% of the total number of positions. A similarity between p35 and M1 was found not only in the amino acid residues sequence, but also in a secondary structure, as far as the matching of alpha-helical segments and loop regions was visualized (Fig. 3). It is of interest that a supermotif region appeared to be identical for all so far sequenced M1 proteins of influenza A virus, and was also found in Bombix mori baculovirus p35 protein. It is important to note that the performed analysis of M1 matrix proteins from influenza B and C viruses did not display a respective similarity with p35 protein, which correlated with the lack of their ability to bind caspase-8 (see above). Despite the evident homology (~39.5%) between the N-terminal primary structures of M1 from influenza A and B viruses (Fig. 3b), a computer search did not reveal a “supersimilarity” (supermotifs) in this region of influenza B virus M1 and baculovirus p35. A similar trend was also observed for influenza C virus M1, proving the lack of the supermotif in M1 N-terminal region, homological with that of p35. In both cases the similarity score in the identified supermotif region was 0.5-1.9, whereas that for M1/A - p35 was 4.4 (Fig. 3c). An analogous comparative study of a number of other apoptosis inhibitors, such as XIAP, IAP [35], usurpin [36], adenovirus protein of 14.7 kD molecular weight [37], smallpox virus crmA protein [38], has not revealed any domains (supermotifs) displaying an appreciable extent of similarity with M1 protein (data not shown).Fig. 2. A comparison of caspase-8 binding by M1 matrix protein from influenza A, B, and, C viruses. Purified preparations of influenza A/Aichi/68 (A), B/Hong Kong/72 (B), C/USSR/77 (C) viruses were subjected to PAGE followed by Coomassie blue treatment (a) and Western blot transfer on the PVDF membrane (b), which was further incubated in a solution with a ligand, caspase-8 (catch overlay assay). Caspase-8 adsorbed on the membrane was identified using anti-caspase antibodies and HRP conjugate by enhanced chemiluminescence method. Lane K shows purified caspase-8 which was used as a ligand. The stars indicate HA2 polypeptide position.
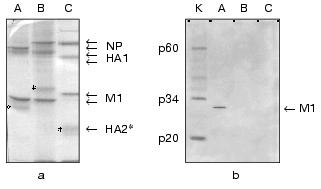
Fig. 3. A comparison of the structures of M1 protein from influenza A, B, and C viruses and p35 baculovirus antiapoptotic protein. A comparison was performed in a pairwise multiple sequence alignment program [26]. Dot, plus, or star symbols above the aligned amino acid pair show the ranges of similarity score: 1SD below the average value of Johnson's matrix (*), or 1SD (+) and 2SD (*) above that one, respectively [26]. a) Partial pairwise alignment of M1 protein from influenza A virus (A/FPV/34 [28]) with p35 protein from Autographa californica [29]. A region containing a homologous supermotif is presented. b) Full pairwise alignment of M1 protein from influenza A virus (FPV) with M1 proteins of influenza B/Lee/40 virus [30] and C/Mississippi/80 [31]. First 90 amino acids of M1 protein from influenza A virus are shown. c) Similarity analysis of amino acid regions 63-75 of p35 protein and 20-32 of M1 protein from influenza viruses of three different types. Coefficients of power (P) illustrating the degree of “non-coincidence” of similarity [26] and a percentage number of positions bearing homologous amino acids (%) in the sequence of analyzed region of the molecule. Primary structures of M1 and p35 proteins are recorded in single letter format. The figures above indicate the amino acid numbers in the analyzed sequences. Homologous region (supermotif) is marked with bold font. alpha-Helix regions are numbered beginning with the N-terminal region and placed within a frame; an arrow shows pentapeptide in p35 (underlined) that is involved in the interaction with caspase catalytic site [32].

DISCUSSION
The present investigation has demonstrated that the main structural protein of influenza A virus, which forms the protein matrix of the virion, is capable of binding caspase-8. The discovered feature of M1 protein is probably controlled by the N-terminal region, displaying a similarity with a fragment of an active site of baculoviral p35 protein, a caspase inhibitor. This conclusion is in good correlation with the previous observations on the presence of protease-binding (PB) domain in the N-terminal region of M1 molecule, and its ability to bind serine proteases [5, 12]. PB domain of M1 protein also resembles a fragment of an active site of aprotinin [12], an inhibitor of a large number of serine proteases [39]. It is quite probable that the N-terminal region of M1 contains a protease-binding domain, responsible for the binding of serine as well as cysteine proteases. Two aspects should be kept in mind in the context of this supposition. First, this idea is confirmed by the data on the lack of caspase-8-binding activity of influenza B and C viruses M1 matrix protein, and earlier observations [12], illustrating the lack of PB domain and hence the ability of the above mentioned M1 proteins to bind serine proteases. Second, cysteine proteases have a profound structural and functional similarity with serine proteases, and proteases of both classes may interact with the natural inhibitors and inhibitor-like proteins in a similar way.
It should be noted that during the apoptosis initiation process caspase-8 undergoes an auto-aggregation and activation [25] via its interaction with an effector DED-domain of the cellular adapter FADD protein molecule, which is associated with apoptosis Fas-receptor [40]. The interesting observation we made from a comparison of the primary structures of FADD DED-domain and influenza A virus M1 matrix protein, which indicated a 35% of homology between the primary structures of M1 N-terminal region (amino acid residues at the positions 25-40 [41]) and amino acid sequence of FADD effector domain (positions 44-67 [42]) (data not shown). This FADD region plays an important role in the proapoptotic aggregation and activation (a signal transduction) of caspase-8 [40]. It can be assumed that M1 can interfere with the interaction process between caspase-8 and FADD, as a result of direct interaction with caspase-8 and owing to its similarity with FADD DED-domain, followed by FADD-dependent auto-aggregation and activation of caspase-8.
Caspase-8 is one of the two main inducers (along with caspase-9) of the biochemical cascade of programmed cell death, so-called “apoptosis” [43]. This fact raises a special interest towards the phenomenon of M1 and caspase-8 interaction, which we are reporting here, since it has been well known that influenza virus causes the apoptosis of the infected cells [16, 17] by a caspase-8-dependent pathway [18]. However, it is still unclear whether the supposed interaction and M1 interference with caspase-8 would have pro- or anti-caspase nature. The data we obtained on the apoptosis inhibition induced by tissue necrosis factor TNF-alpha [21], which is known to be developed in the human carcinoma cells Hep-2 expressing M1 protein by a receptor caspase-8-dependent pathway, provide evidence for an antiapoptotic function of influenza A virus M1 protein. It is not inconceivable that in the antiapoptotic program M1 protein is functioning in cooperation with NS1 viral protein, which has an antiapoptotic potential [20]. In order to evaluate caspase recognition and binding, we are planning experiments with M1 protein, with its N-terminal PB domain altered by a site-specific mutagenesis, which will define the role of M1 viral protein in influenza-induced cellular apoptosis.
The authors are grateful to G. Salvesen (The Burnham Institute, CA, USA) for providing us with the purified samples of caspases-3, -6, -7, and -8. This work was supported by the Russian Foundation of Basic Research (RFBR) (grant Nos. 04-48442 and 04-04000), RFBR/DFG 436113/587, Howard Hughes Institute (USA) (grant No. 75195-546302), and a grant of NATO scientific collaboration HT-974619.
REFERENCES
1.Schulze, I. (1972) Virology, 47,
181-196.
2.Gregoriades, A., and Frangione, B. (1981) J.
Virol., 40, 323-328.
3.Fujioshi, Y., Kume, N. P., Sakata, K., and Sato, S.
B. (1994) EMBO J., 13, 318-326.
4.Ruigrok, R. W. H., Barge, A., Durre, P., Brunner,
J., Ma, K., and Whittaker, R. G. (2000) Virology, 267,
289-298.
5.Timofeeva, T. A., Klenk, N. D., and Zhirnov, O. P.
(2001) Mol. Biol. (Moscow), 35, 484-491.
6.Whittaker, G., Bui, M., and Helenius, A. (1996)
Trends Cell Biol., 6, 67-71.
7.Avalos, R. T., Yu, Z., and Nayak, D. P. (1997)
J. Virol., 71, 2947-2958.
8.Gomez-Puertas, P., Albo, C., Perez-Pastrana, E.,
Vivo, A., and Portela, A. (2000) J. Virol., 74,
11538-11547.
9.Ali, A., Avalos, R. T., Ponimaskin, E., and Nayak,
D. P. (2000) J. Virol., 74, 8709-8719.
10.Ye, Z., Baylor, N. W., and Wagner, R. R. (1989)
J. Virol., 63, 3586-3594.
11.Zhirnov, O. P., and Klenk, H. D. (1997)
Virology, 235, 302-310.
12.Zhirnov, O. P., Ksenofontov, A. L., and Klenk, N.
D. (1999) Dokl. RAN, 367, 690-693.
13.Elster, C., Larsen, K., Gagnon, J., Ruigrok, R.
W. H., and Baudin, F. (1997) J. Gen. Virol., 78,
1589-1596.
14.Ye, Zh., Robinson, D., and Wagner, R. R. (1995)
J. Virol., 69, 1964-1970.
15.Rey, O., and Nayak, D. P. (1992) J.
Virol., 66, 5815-5824.
16.Takizawa, T., Matsukawa, S., Higuchi, Y.,
Nakamura, S., Nakanishi, Y., and Fukuda, R. (1993) J. Gen.
Virol., 74, 2347-2355.
17.Hinshaw, V. G., Olsen, C. W., Dybgahi-Sissoko,
N., and Evans, D. (1994) J. Virol., 68, 3667-3673.
18.Balachandran, S., Roberts, P. C., Kipperman, T.,
Bhalla, K. N., Compans, R. W., Archer, D. R., and Barber, G. N. (2000)
J. Virol., 74, 1513-1523.
19.Schultz-Cherry, S., Dybdahl-Sissoko, N., Neumann,
G., Kawaoka, Y., and Hinshaw, V. S. (2001) J. Virol., 75,
7875-7881.
20.Zhirnov, O. P., Konakova, T. E., Wolff, T., and
Klenk, H. D. (2002) J. Virol., 76, 1617-1625.
21.Zhirnov, O. P., Konakova, T. E., Krichevets, S.
G., Iordansky, S. N., and Klenk, H. D. (2000) Abst. XI Int. Conf.
Negative Strand Viruses, Quebec, Canada, p. 91.
22.Zhirnov, P., Ovcharenko, A. V., and Bukrinskaya,
A. G. (1985) J. Gen. Virol., 66, 1633-1638.
23.Zhirnov, O. P. (1992) Virology,
186, 324-330.
24.Kyhse-Anderson, J. (1984) J. Biochem. Biophys.
Meth., 10, 203-209.
25.Stennicke, H. R., and Salvesen, G. S. (2000)
Biochim. Biophys. Acta, 1477, 299-306.
26.Leontovich, A. M., Brodsky, L. I., and
Golbalenya, A. E. (1993) Biosystems, 30, 57-63.
27.Johnson, M. S., and Overington, J. P. (1993)
J. Mol. Biol., 233, 716-738.
28.McCauley, J. W., Mahy, B. W., and Inglis, S. C.
(1982) J. Gen. Virol., 58, 211-215.
29.Friesen, P. D., and Miller, L. K. (1987) J.
Virol., 61, 2264-2272.
30.Briedis, D. J., Lamb, R. A., and Choppin, P. W.
(1982) Virology, 116, 581-588.
31.Tada, Y., Hongo, S., Muraki, Y., Sugawara, K.,
Kitame, F., and Nakamura, K. (1997) Virus Genes, 15,
53-59.
32.Zhou, Q., Krebs, J. F., Snipas, S. J., Price, A.,
Alnemetri, E. S., Tomaselli, K. J., and Salvesen, G. S. (1998)
Biochemistry, 37, 10757-10765.
33.Bertin, J., Mendrysa, S. M., LaCount, D. J.,
Gaur, S., Krebs, J. F., Armstrong, R. C., Tomaselli, K. J., and
Friesen, P. D. (1996) J. Virol., 70, 6251-6259.
34.Fisher, A. J., Cruz, W., Zoog, S. F., Schneider,
C. L., and Friesen, P. D. (1999) EMBO J., 18,
2031-2039.
35.Deveraux, Q. L., Leo, E., Stennicke, H. R.,
Welsh, K., Salvesen, G. S., and Reed, J. C. (1999) EMBO J.,
18, 5242-5251.
36.Rasper, D. M., Vaillancourt, J. P., Hadano, S.,
et al. (1998) Cell Death Differ., 5, 271-288.
37.Tufariello, J. M., Cho, S., and Horwitz, M. S.
(1994) Proc. Natl. Acad. Sci. USA, 91, 10987-10991.
38.Zhou, Q., Snipas, S., Orth, K., Muzio, M., Dixit,
V. M., and Salvesen, G. S. (1997) J. Biol. Chem., 272,
7797-7800.
39.Fritz, H., and Wunderer, G. (1983) Drug
Res., 33, 479-494.
40.Berglund, H., Olerenshaw, D., Sankar, A.,
Federwisch, M., McDonald, N. Q., and Driscoll, P. C. (2000) J. Mol.
Biol., 302, 171-188.
41.McCauley, J. W., Mahy, B. W. J., and Inglis, S.
G. (1982) J. Gen. Virol., 58, 211-215.
42.Boldin, M. P., Varfolomeev, E. E., Pancer, Z.,
Mett, I. L., Camonis, J. H., and Wallach, D. (1995) J. Biol.
Chem., 270, 7795-7798.
43.Green, D. G. (1998) Cell, 94,
695-698.