Interaction between Tubulin and Na+,K+-ATPase in Brain Stem Neurons
N. M. Vladimirova*, E. N. Sautkina, T. V. Ovchinnikova, and N. A. Potapenko
Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117997 Russia; fax: (095) 336-4444; E-mail: vla@mail.ibch.ru* To whom correspondence should be addressed.
Received November 8, 2001; Revision received December 5, 2001
Functionally active Na+,K+-ATPase isozymes containing three types of the catalytic subunits (alpha1, alpha2, and alpha3) were obtained from calf brain by two methods: selective removal of contaminating proteins according to Jorgensen (1974) and selective solubilization of the enzyme with subsequent reformation of the membrane structure according to Esmann (1988). All preparations were characterized with respect to ouabain-inhibition constants. The presence of the cytoskeleton protein tubulin (beta3 isoform) in the high-molecular-weight complex of Na+,K+-ATPase alpha3beta1 isozyme from brain stem axolemma and the junction between Na+,K+-ATPase alpha3 subunit and tubulin beta3 subunit are shown for the first time.
KEY WORDS: Na+,K+-ATPase, isoforms, isozymes, axolemma, tubulin
Na+,K+-ATPase (Na+-pump, EC 3.6.1.37) is a membrane-bound oligomer consisting of equimolar amounts of catalytic (alpha) and glycosylated (beta) subunits. The existence of four isoforms of subunit alpha (alpha1, alpha2, alpha3, and alpha4) and three isoforms of subunit beta (beta1, beta2, and beta3), which are the products of different genes, was shown [1-3]. According to current notion on Na+-pump structure, this enzyme is not represented solely by the well-described heterodimer alpha1beta1 (the “kidney” type), but is a family of 12 potentially possible types of isozymes with different alphanbetam heterodimeric structure. The situation with the number of isozymes is complicated by the fact that is some tissues Na+,K+-ATPase complex is associated with the third subunit gamma, whose assignment and function are widely debated in literature [2, 3]. It has been shown recently that the amount and distribution of the isoforms is characterized by tissue and cell specificity, is fairly determined in the cells with complex organization, and changes during ontogeny and in pathologies [2, 3]. The function of Na+,K+-ATPase in the brain is poorly studied and least understood. It was shown that in the brain, in addition to the conventional functions, Na+,K+-ATPase is involved in intercellular interaction between the neurons and glial cell [4]. In addition, it was shown that this enzyme is associated with the cytoskeletal proteins, where all known isoforms of both subunits are expressed [5, 6].
The alpha1 isoform of the catalytic subunit has received the most study, and its structure was determined for the majority of animals. It is believed that alpha1 isoform regulates the cytosolic Na+ content. It is expressed in almost all tissues and is called a housekeeping isoform. Practically all data on the topography and functional domains of Na+,K+-ATPase were obtained using the alpha1beta1 isozyme. The structure of alpha2 and alpha3 isoforms was determined for a small number of objects; the distribution of these isoforms is more tissue-specific. The alpha4 isoform was isolated from the only source (rat testis), although a small amount of this isoform was also detected in the brain [3]. The functional properties of alpha4 isoform remain obscure.
Despite the fact that the three isoforms of the catalytic subunit (alpha1, alpha2, and alpha3) share significant structural homology (more than 94%), they display significant functional differences. For instance, they are differentially inhibited by cardiotonic plant steroids (e.g., ouabain) [1, 7, 8] and endogenous digitalis-like compounds as well [3, 9] and markedly differ with respect to antagonism between K+ and ouabain at physiological K+ concentrations [3, 7]. In addition, these isoforms differ in their affinity for ATP and Na+, sensitivity to Ca2+ inhibition, resistance to oxidants, radicals, and endogenous proteinases, as well as in the mechanisms of regulation of their biosynthesis and activity [9]. The reason for these prominent differences is not clear; the results published in literature are contradictory. The data on the functional properties of isoforms were obtained mostly by analyzing either the microsomes containing these isoforms or the products of expression of the corresponding genes encoding the subunits rather than by studying the isolated isoforms and isozymes. The analysis of the data published in literature showed that the broad spectrum of functional differences between the isoforms cannot be due solely to the fine differences in their primary structure. These differences may be also caused by lipid environment [2, 3], the carbohydrate component of the beta subunit [10], and their interaction with membrane and cytoplasmic proteins [2, 3, 6]. Therefore, the system used for assaying the properties of isoforms and isozymes should be maximally close to the in vivo conditions.
Na+,K+-ATPase is usually isolated using two different methodical approaches: a) selective removal of contaminating proteins using the ionic detergent sodium dodecyl sulfate (SDS) in the presence of ATP (the method of Jorgensen [11]), when Na+,K+-ATPase remains membrane-bound, functionally active, in native lipid environment, and b) selective solubilization of the catalytically active Na+,K+-ATPase using nonionic detergent C12E8 (octaethylene glycol dodecyl ether) with subsequent reconstruction of the membrane structure in the presence of Ca2+ or Mn2+ (the method of Esmann [12]). Given that this detergent allows sufficient retaining the protein-protein contacts, it was widely used previously to study the subunit structure of many membrane proteins and their interaction with external protein structures.
The method of Jorgensen has been the main procedure used for isolating Na+,K+-ATPase from the kidneys of different animals: rabbit, dog, sheep [13], pig [13, 14], calf [10], as well as from duck salt glands [15], which contain isozymes of the alpha1beta1 type. In these preparations, the alpha subunit accounts for 67-70% protein. We also demonstrated the absence of contaminating proteins when analyzing the peptides obtained by exhaustive hydrolysis of exposed domains of the membrane-bound enzyme from pig kidneys: the hydrolyzate contained peptides of only the alpha1 and beta1 subunits of Na+,K+-ATPase [14]. However, when using this procedure for isolation of isozymes comprised of other types of the catalytic subunit, researchers ran into some obstacles. For example, Na+,K+-ATPase preparation isolated from the axolemma (plasma membrane of myelinated axons) of rat brain stem, which, as determined by immunochemical analysis, contain a mixture of the alpha2 and alpha3 isoforms, with respect to enzymatic activity is comparable with the enzymes isolated from kidneys [16]. However, the results of electrophoresis showed that, unlike kidney Na+,K+-ATPase, even the purest axolemmal Na+,K+-ATPase preparations obtained by the method of Jorgensen contained in the zone of 50-kD proteins, in addition to beta subunit, a contaminating protein with a similar molecular weight, which was not identified. The success of Na+,K+-ATPase isolation from membrane homogenate depended on ATPase activity of the microsomal fraction and the way it was obtained. The final isoform composition of Na+,K+-ATPase changed with the isolation procedure and the object of study. For example, axolemmal Na+,K+-ATPase preparations isolated from the brain stem of the dog, in contrast to analogous preparation isolated from the rat, contained not only the alpha2 and alpha3 isoforms, but the alpha1 isoform as well. The analysis of Na+,K+-ATPase preparations isolated by the method of Jorgensen from shrimp cysts [17] (containing catalytic subunit isoforms similar in structure to the mammalian alpha3 isoform) and shark rectal glands [12] (which presumably contain the alpha3 isoform [18]) revealed the presence of additional proteins (whose structure, unfortunately, was not analyzed). For further purification of Na+,K+-ATPase preparations isolated from shark rectal glands, Esmann applied selective solubilization of the enzyme with the use of detergent C12E8 with subsequent reconstitution of the membrane structure in the presence of Ca2+ or Mn2+ [12].
In our recent studies, we analyzed the structure of Na+,K+-ATPase isolated from calf brain [5, 6]. We determined the N-terminal sequences of the alpha1, alpha2, alpha3, beta1, and beta2 isoforms and analyzed the isozyme composition and type of functioning alphanbetam complexes in the brain parts containing different types of cells. It is known from the literature that the properties of highly purified preparations containing identical Na+-pump isoforms are not only tissue-specific, but also depend on the protocol of enzyme purification. For this reason, for the isolation of Na+,K+-ATPase from calf brain we used both common methods (the methods of Jorgensen and Esmann).
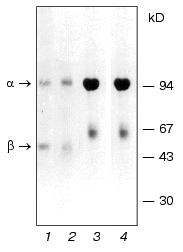
The enzyme preparations isolated from gray matter by the two above-described methods had comparable ATPase activity, characteristic biphasic kinetics of ouabain inhibition (Ki ~ 10-6 M and 1.5*10-8 M), and a similar set of proteins (as judged by the results of electrophoretic and structural analysis). It is shown that Na+,K+-ATPase from gray matter containing glial cells represents a set of isozymes of alpha1beta1, alpha2beta2, and alpha3beta1 types, with the alpha1beta1 isoform being predominant. It is known that beta2 isoform is homologous to the protein that was previously known as the “adhesion molecule on the glia”. This protein ensures the interaction between astrocytes and neurons in the nervous system. The structure of its polysaccharide chains is similar to that of the complex glycans in adhesion molecules [4]. Because the content of beta2 isoform in the gray matter is comparable to that of alpha2 isoform, it can be assumed that Na+,K+-ATPase isozyme alpha2beta2 is a candidate for the role of adhesion molecule. Comparative electrophoretic analysis of the enzyme preparations isolated from the gray substance and kidney external medullar layer of the calf showed that brain beta1 subunit displays greater electrophoretic mobility (Fig. 1). The analysis of the carbohydrate component showed that Na+,K+-ATPase preparations of similar isozyme composition obtained from the brain and kidneys differ in the extent of glycosylation. We discovered that brain beta1 subunit is characterized by a simpler structure of the carbohydrate chain [10], which may be a reason for differential resistance of these enzymes to endogenous proteinases and free radicals.
It was shown earlier that Na+,K+-ATPase from calf brain stem neurons and axolemma is a mixture of alpha2beta1 and alpha3beta1 isozymes with equal sensitivity to ouabain (Ki ~10-7 M), with the alpha3beta1 type being predominant in axolemma [5]. When isolating and analyzing Na+,K+-ATPase preparations from the brain stem neurons and axolemma, we ran into some problems. First, the alpha2-to-alpha3 ratio and the presence of additional protein bands depended not only on the microsomal fraction, but also on the protocol of enzyme isolation and purification. We discovered that one of the reasons explaining this phenomenon was an increased sensitivity of the catalytic subunit alpha3 to endogenous proteinases resulting in proteolysis of the subunit and formation of the N-terminal fragment with the molecular weight of approximately 55 kD, which is visualized as a band in the “beta-subunit zone” [5]. We also identified the site for trypsin-like endogenous proteolysis in the polypeptide chain unique for the alpha3 subunit: -Pro-Asn-Asp-Asn-Arg492 v (Tyr493-Leu-Leu-Val-Met). Treatment with the proteinase inhibitor diisopropyl fluorophosphate (DIFP) prior to detergent addition was required both at the stage of enzyme isolation and electrophoretic analysis. However, even after pretreatment with the inhibitor, an additional protein band was still detected (the upper band of the “beta-subunit zone”, Fig. 2, lane 2), which was similar to the protein band detected by Sweadner in Na+,K+-ATPase preparations isolated from rat brain stem [16]. We discovered that the intensity of this protein band correlated with the content of Na+,K+-ATPase alpha3 isoform. Structural analysis showed that this band is the beta isoform of tubulin.Fig. 1. Electrophoregram of Na+,K+-ATPase preparations from gray matter of calf brain cortex (1, 2) and medullar layer of calf kidney (3, 4) treated (2, 4) or not treated (1, 3) with 2-mercaptoethanol. The specimens were analyzed by SDS-PAGE by the method of Weber and Osborn [19] in gradient (4-15%) polyacrylamide gel in 0.1 M sodium phosphate buffer (pH 7.2) containing 0.1% SDS at 15°C. Before the addition of 2-mercaptoethanol and SDS (to the final concentration of 2.5 and 5%, respectively), the specimens were incubated for 15 min at room temperature in buffer containing 3 mM DIFP and 1 mM EDTA. The gel was stained with Coomassie Brilliant Blue G-250.
As mentioned above, axolemma is most enriched in the alpha3 isoform of Na+,K+-ATPase. The procedure of axolemma isolation from the brain stem involved isolating myelinated axons with subsequent dissociation and segregation of myelin from the axolemma [20]. Gradient centrifugation allowed us to segregate two axolemmal fractions of different density, each of which was then used for isolating Na+,K+-ATPase by the methods of Jorgensen and Esmann. Subsequent analysis of the enzyme structure showed that the preparation obtained from the axolemmal fraction of higher density using both methods contained predominantly the alpha3 and beta1 subunits of Na+,K+-ATPase and beta isoform of tubulin. This is the first time when a protein complex of such composition was isolated. In view of this, the questions emerge as to which isoform of beta tubulin is present in the complex, whether Na+,K+-ATPase is linked to tubulin, and, if it is, which Na+,K+-ATPase subunit (alpha3 or beta1) is linked with tubulin and of what nature is this linkage. Isoforms of alpha and beta subunits of tubulin are representative of highly homologous protein families. For example, beta isoforms of tubulin share more than 95% homology. It is known that expression and modification of alpha and beta subunit of tubulin are especially complicated in the brain: up to nine different but highly homologous isoforms of beta tubulin, whose biological role remains obscure, are expressed here [21].Fig. 2. Electrophoregram of Na+,K+-ATPase preparations from calf brain stem axolemma treated (2) or not treated (1) with 2-mercaptoethanol. The standard mixture of molecular weight markers is shown in lane 3. The arrows show the proteins whose structure was then analyzed as described in the text. The specimens were subjected to electrophoresis and treated as described in the legend to Fig. 1. One half of the gel was stained with Coomassie Brilliant Blue G-250. The other (unstained) half of the gel containing the same proteins was analyzed by electroblotting on PVDF membrane in 0.025 M sodium bicarbonate buffer (pH 9.0) containing 0.1% SDS and 20% methanol. The protein bands on the membrane were detected either by staining with amido black 10B or visually (without staining) in the course of membrane drying as described in [6]. The cut-out bands strips of PVDF membrane containing protein bands were then used for sequencing and hydrolysis of C-terminal amino acids with carboxypeptidases.

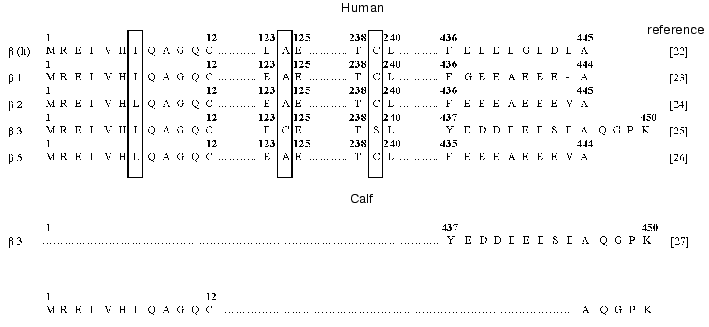
The results of sequencing of twelve N-terminal amino acid residues of tubulin were insufficient to identify the isoform because of the high extent of homology between beta isoforms in this domain (Fig. 3). For this reason, we additionally performed step-wise hydrolysis of the C-terminal amino acid residues with the use of carboxypeptidases (cpB + cpA, pH 8.5, 1 h; cpA + cpY, pH 7.0, 1 h). As a result, we determined the sequence of five C-terminal amino acid residues of tubulin (Fig. 3). The comparison of the sequences determined with the known primary structures of human beta tubulins [22-26] and C-terminal sequence of calf beta3 isoform [27] showed that the Na+,K+-ATPase preparation contained the beta3 isoform of tubulin.
The beta3 isoform of tubulin was detected in Na+,K+-ATPase preparations isolated both by the method of Jorgensen and the method of Esmann, which are based on different principles of isolation (i.e., obtaining the enzyme in the membrane-bound and solubilized forms, respectively) and the use of different detergents (the ionic detergent sodium dodecyl sulfate and nonionic detergent C12E8, respectively). This is suggestive of possible linkage between the Na-pump and cytoskeletal protein tubulin.Fig. 3. Comparative analysis of the fragments of amino acid sequences of beta isoforms of human and calf tubulin. The N- and C-terminal amino acid sequences of tubulin contained in the Na+,K+-ATPase preparation from calf brain stem axolemma, which were determined in this study, are shown at the bottom of the scheme.
Electrophoretic analysis yielded some interesting findings: the electrophoregrams of Na+,K+-ATPase isolated from gray matter and kidney treated and not treated with 2-mercaptoethanol (Fig. 1) are similar: the electrophoregrams show two protein bands corresponding to the alpha and beta subunits of Na+,K+-ATPase. This is indicative of the absence of disulfide bonds of alpha-alpha, alpha-beta, and beta-beta type. The electrophoregrams of enzymes isolated from the axolemma are different from electrophoregrams of enzyme obtained from the kidneys and gray matter. First, in preparations treated with 2-mercaptoethanol, three protein bands corresponding to the alpha3 and beta1 subunits of Na+,K+-ATPase and beta3 subunit of tubulin are detected (Fig. 2, lane 2). Second, the preparations that were not treated with 2-mercaptoethanol lack the bands corresponding to the beta3 isoform of tubulin and alpha3 subunit of Na+,K+-ATPase. However, they contain a new band near the origin (Fig. 2, lane 1). The analysis of N-terminal sequence of the protein band located near the origin showed the presence of an equimolar mixture of two amino acid sequences corresponding to the alpha3 isoform of Na+,K+-ATPase and beta3 isoform of tubulin.
The data obtained in this study led us to assume that the alpha3 subunit of Na+,K+-ATPase may be linked to the beta3 subunit of tubulin via a disulfide bond.
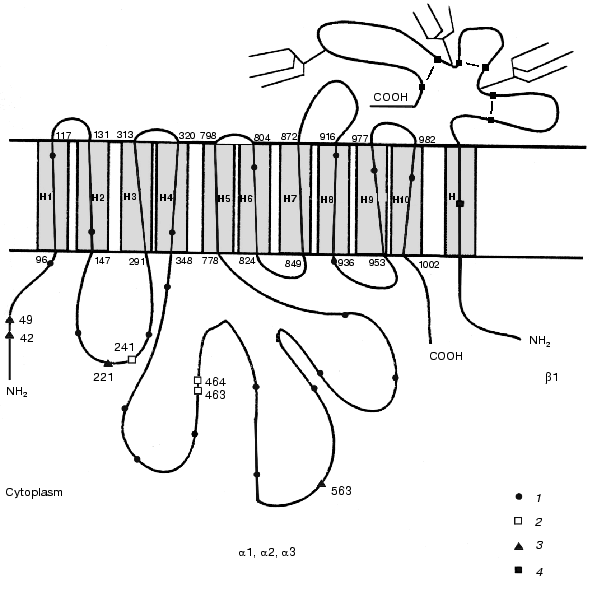
The comparison of all known primary structures of alpha subunit isoforms revealed an interesting structural peculiarity of the alpha3 isoform. Besides the twenty-one conserved cysteine residues characteristic of all isoforms of the catalytic subunit, the alpha3 isoforms (to date determined for the rat, human, and chick) additionally contain four cysteine residues (Cys42, Cys49, Cys221, and Cys563) (the numeration is given according to rat kidney Na+,K+-ATPase sequence), which are located in the cytoplasmic domain. Conversely, the alpha3 isoform lacks Cys463 and Cys464, which are characteristic of the alpha1 isoform, or Cys241 and Cys464, which are characteristic of the alpha2 isoform (Fig. 4) and are also located in the cytoplasmic domain. However, the alpha1, alpha2, and alpha3 isoforms of the catalytic subunit differ not only in the total content of sulfhydryl groups, but also in their reactivity. For example, Sweadner showed that the number of reactive sulfhydryl groups in the preparations of brain ouabain-sensitive catalytic subunit alpha+ (the mixture of the alpha2 and alpha3 isoforms) involved in the reaction with N-ethylmaleimide is twofold greater than that in the preparations of kidney catalytic subunit alpha1 [29]. Esmann showed that Na+,K+-ATPase preparations from shark rectal glands and kidneys (containing the alpha3 and alpha1 isoforms of the catalytic subunit, respectively) are differentially inactivated by N-ethylmaleimide [30].
Comparison of amino acid sequences of different tubulin isoforms also revealed an interesting characteristic of beta3 isoform: the positions of cysteine residues in this isoform differ from those in other isoforms. Note that the positions of cysteine residues in different isoforms of beta subunit of tubulin are fairly conserved; however, the beta3 isoform differs from the other tubulin isoforms by the absence of Cys239 and presence of an additional residue Cys124 (Fig. 3).Fig. 4. Positions of cysteine residues in alpha1, alpha2, alpha3, and beta1 isoforms of Na+,K+-ATPase: 1) cysteine residues in the alpha3 isoform homologous to those in the alpha1 and alpha2 isoforms; 2) cysteine residues that are absent from the alpha3 isoform; 3) cysteine residues that are present only in the alpha3 isoform; 4) cysteine residues homologous to those in beta1 isoforms of different species. The topographic model of the enzyme shown in the figure was proposed by Karlish et al. [28].
The data led us to conclude that both the alpha3 isoform of Na+,K+-ATPase and the beta3 isoform of tubulin contain additional cysteine residues characteristic of these types of isoforms. The possibility of formation of a specific disulfide bond between the alpha3 isoform of Na+,K+-ATPase and beta3 isoform of tubulin exists.
The physiological role of association between the Na-pump and tubulin isoforms remains obscure. It is known, however, that other cytoskeletal proteins (actin, fodrin, and ankyrin/spectrin) interact with Na+,K+-ATPase. It was shown that 125I-labeled ankyrin interacts with the alpha1 subunit of Na+,K+-ATPase from the MDCK cell line [31] and that this interaction is noncovalent [32]. The domains of the polypeptide chain of the alpha subunit involved in the interaction with ankyrin were determined. These domains are highly conserved in all Na+,K+-ATPase isoforms [33].
It was shown that in epithelial cells and arterial muscle cells (which, similarly to neurons, display complex organization) the polarized distribution of Na+,K+-ATPase is ensured by its interaction with the cytoskeletal proteins spectrin and ankyrin [34, 35]. Using cytoimmunochemical technique, it was shown that alpha2 isoform in the astrocytes and alpha3 isoform in neurons and myocytes are located near the plasma membrane region that is parallel to endoplasmic/sarcoplasmic reticulum, where the Na-pump is also contained. It was assumed that alpha1 isoform regulates total Na+ concentration and, indirectly, Ca2+ concentration, whereas alpha2 and alpha3 isoforms regulate the concentration of these ions in the restricted space between the plasma membrane and endoplasmic/sarcoplasmic reticulum [36]. It has been recently reported that tubulin dimer (or subunit) is anchored in the ganglioside-rich domains of neurons, which are resistant to detergents. This anchored tubulin plays a key role in forming junctions between the microtubules and plasma membranes required for membrane remodeling in the course of axon growth. Based on these findings, it was assumed that tubulin possibly interacts with the protein components of the plasma membrane [37].
Thus, it is evident that the functional properties and characteristics of Na+,K+-ATPase isozymes are determined not only by the isozyme composition, lipid environment, and differences in glycosylation, but also by association of the enzyme with the cytoskeletal proteins and other protein components.
This study was supported by the Russian Foundation for Basic Research (project No. 99-04-48387).
REFERENCES
1.Sweadner, K. J. (1985) J. Biol. Chem.,
260, 11508-11513.
2.Mobasheri, A., Avila, J., Cozar-Castellano, I.,
Brownleader, M. D., Trevan, M., Francis, M. J. O., Lamb, J. F., and
Martin-Vasallo, P. (2000) Biosci. Reports, 20, 51-91.
3.Blanco, G., and Mercer, R. W. (1998) Am. J.
Physiol. Renal Physiol., 275, F633-F650.
4.Gloor, S., Antonicek, H., Sweadner, K. J.,
Pagliusi, S., Frank, R., Moos, M., and Schachner, M. (1990) J. Cell.
Biol., 110, 165-174.
5.Vladimirova, N. M., Platoshkina, E. A., Efendiyev,
R. E., and Potapenko, N. A. (1997) Ann. N. Y. Acad. Sci.,
834, 153-154.
6.Vladimirova, N. M., Potapenko, N. A., and Modyanov,
N. N. (1995) Bioorg. Khim., 21, 415-422.
7.Crambert, G., Hasler, U., Beggah, A. T., Modyanov,
N. N., Horisberger, J., Lelievre, L., and Geering, K. (2000) J.
Biol. Chem., 275, 1976-1986.
8.Maixent, J. M., Lelievre, L., and Berrebi-Bertrand,
I. (1988) Cardiovasc. Drugs Ther., 12, 585-594.
9.Therien, A. G., and Blostein, R. (2000) Am. J.
Physiol. Cell Physiol., 279, C541-C566.
10.Vladimirova, N. M., Murav'eva, T. I.,
Ovchinnikova, T. V., Potapenko, N. A., and Khodova, O. M. (1998)
Membr. Cell. Biol., 12, 435-439.
11.Jorgensen, P. L. (1974) Biochim. Biophys.
Acta, 356, 36-52.
12.Esmann, M. (1988) Biochim. Biophys. Acta,
940, 71-76.
13.Jorgensen, P. L. (1988) Meth. Enzymol.,
156, 29-43.
14.Ovchinnikov, Yu. A., Arzamazova (Vladimirova), N.
M., Arystarkhova, E. A., Gevondyan, N. M., Aldanova, N. A., and
Modyanov, N. N. (1987) FEBS Lett.,217,269-274.
15.Smith, T. W. (1988) Meth. Enzymol.,
156,46-48.
16.Sweadner, K. J. (1988) Meth. Enzymol.,
156, 65-76.
17.Peterson, G. L., and Hokin, L. E. (1988) Meth.
Enzymol., 156, 48-65.
18.Fisone, G., Cheng, S. X., Nairn A. C., Czernik,
A. J., Hemmings, H. C., Hoog, J.-O., Bertorello, A. M., Kaiser, R.,
Bergman, T., Jornvall, H., Aperia, A., and Greengard, P. (1994) J.
Biol. Chem.,269,9368-9373.
19.Weber, K., and Osborn, M. (1969) J. Biol.
Chem., 244,4406-4412.
20.De Vries, G. H., Matthieu, Y.-M., Beny, M.,
Chicheportiche, R., Lazdunski, M., and Dolivo, M. (1978) Brain
Res., 147, 339-352.
21.MacRae, T. H. (1992) Biochem. Cell. Biol.,
70, 835-841.
22.Leffers, H., Wiemann, S., and Ansorge, W. (1994)
submitted to the Protein Sequence Database.
23.Lee, M. G., Lewis, S. A., Wild, C. D., and Cowan,
N. J. (1983) Cell, 33, 477-487.
24.Lewis, S. A., Gilmartin, M. E., Hall, J. L., and
Cowan, N. J. (1985) J. Mol. Biol., 182, 11-20.
25.Randanathan, S., Dexter, D. W., Benetatos, C. A.,
and Hudes, G. R. (1998) Biochim. Biophys. Acta, 1395,
237-245.
26.Lee, M. G., Loomis, C., and Cowan, N. J. (1984)
Nucleic Acids Res., 12, 5823-5836.
27.Alexander, J. E., Hunt, D. F., Lee, M. K.,
Shabanowitz, J., Michel, H., Berlin, S. C., MacDonald, T. L., Sundberg,
R. J., Rebhun, L. I., and Frankfurter, A. (1991) Proc. Natl. Acad.
Sci. USA, 88, 4685-4689.
28.Karlish, S. J. D., Goldshleger, R., and Stein, W.
D. (1990) Proc. Natl. Acad. Sci. USA, 87,
4566-4570.
29.Sweadner, K. J. (1979) J. Biol. Chem.,
254, 6060-6067.
30.Esmann, M. (1982) Biochim. Biophys. Acta,
688, 251-259.
31.Morrow, J. S., Gianci, C. D., Ardito, T., Mann,
A. S., and Kashgarian, M. (1989) J. Cell. Biol., 108,
455-465.
32.Koob, R., Kraemer, D., Trippe, G., Aebi, U., and
Drenckhahn, D. (1990) Eur. J. Cell. Biol., 53,
93-100.
33.Devarajan, P., Scaramuzzino, D. A., and Morrow,
J. S. (1994) Proc. Natl. Acad. Sci. USA, 91,
2965-2969.
34.Nelson, W. J., and Hammerton, R. W. (1989) J.
Cell. Biol., 108, 893-902.
35.Lee, D., Chen, X., and Smith, P. R. (1996)
FASEB J., 10, A 651.
36.Juhaszova, M., and Blaustein, M. P. (1997)
Proc. Natl. Acad. Sci. USA, 94, 1800-1805.
37.Palestini, P., Pitto, M., Tedeschi, G.,
Ferraretto, A., Parenti, M., Brunner, J., and Masserini, M. (2000)
J. Biol. Chem., 275, 9978-9985.