Derivatives of Benzotetrazine-1,3-dioxide Are New NO-donors, Activators of Soluble Guanylate Cyclase, and Inhibitors of Platelet Aggregation
N. V. Pyatakova1, Yu. V. Khropov2, A. M. Churakov3, N. I. Tarasova4, V. A. Serezhenkov4, A. F. Vanin4, V. A. Tartakovsky3, and I. S. Severina1*
1Institute of Biomedical Chemistry, Russian Academy of Medical Sciences, Pogodinskaya ul. 10, Moscow, 119992 Russia; fax: (095) 245-0857; E-mail: khropov_y@pochtamt.ru2Department of Bioorganic Chemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-2788
3Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky pr. 47, Moscow, 117913 Russia; E-mail: churakov@carc.ioc.ras.ru
4Institute of Chemical Physics, Russian Academy of Sciences, ul. Kosygina 4, Moscow, 119991 Russia; E-mail: mikoyan@center.chph.ras.ru
* To whom correspondence should be addressed.
Received October 29, 2001
The ability of 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides to generate nitric oxide (NO) and activate soluble guanylate cyclase was investigated. All of these compounds were found to be thiol-dependent NO-donors and guanylate cyclase activators. The maximal stimulatory effect of 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides was observed at 10 µM concentration and the activity increase was 4.5-, 15.0-, and 8.2-fold in the presence of 20 µM dithiothreitol and 11.3-, 31.6-, and 20.5-fold, respectively, in the presence of added glutathione (100 µM). The NO-dependent mechanism of benzotetrazine-1,3-dioxide nitroderivative-induced activation of soluble guanylate cyclase (in the presence of 100 µM glutathione) was confirmed by the inhibition (by 78%) of 7-nitrobenzotetrazine-1,3-dioxide (10 µM)-stimulated guanylate cyclase activity in the presence of the NO-scavenger 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (Carboxy-PTIO, 50 µM) and by the inhibition with 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 0.3 µM) of 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides (10 µM)-stimulated guanylate cyclase by 34, 69, and 39%, respectively. All compounds used inhibited ADP-induced aggregation of human platelets with IC50 of 10.0, 1.3, and 2.0 µM for 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides, respectively. A clearly defined correlation was established between the ability of the compounds to generate NO, activate soluble guanylate cyclase, and inhibit platelet aggregation.
KEY WORDS: soluble guanylate cyclase, nitric oxide, derivatives of benzotetrazine-1,3-dioxide, platelet aggregation
Nitric oxide (NO) is a potent physiological regulator of the cardiovascular, immune, and nervous systems [1]. Endogenous NO formed from L-arginine under the action of NO-synthase [2] plays a key role in maintaining the normal tone of vascular smooth muscles. This influences systemic blood pressure. NO is presently considered as an endogenous vasodilator and effective regulator of homeostasis. Anti-hypertensive and anti-aggregatory properties of NO are directly associated with the functioning of soluble guanylate cyclase.
Guanylate cyclase (EC 4.6.1.2, guanosine-5´-triphosphate-pyrophosphate lyase (cyclizing)) catalyzes the biosynthesis of guanosine-3´,5´-monophosphate (cGMP), the second messenger, acting as a universal regulator of intracellular metabolism [3].
It has been established that the interaction of NO with the guanylate cyclase heme iron and subsequent nitrosyl-heme complex formation [4] are accompanied by a conformational changes in the guanylate cyclase active center. These changes lead to the increase in the enzyme activity, cGMP elevation in cells, and activation of cGMP-dependent protein kinase (particularly, of protein kinase G . isoform in platelets [5])--which in turn through phosphorylation of a number of endogenous substrates prevents platelet aggregation [6-8].
Nitric oxide formation was also demonstrated to occur due to biotransformation of nitroglycerin and other nitrates used in treatment of cardiovascular diseases as anti-hypertensive and anti-anginal pharmaceuticals. However, all of them have essential drawbacks: these are weak anti-aggregatory activity, induction of nitrate tolerance as a result of chronic continuous nitrate therapy; emergence and development of other side effects [9]. In view of this, an intensive search for new compounds capable of generating NO nonenzymatically in the living organism (e.g., spontaneously or in a thiol-dependent way) is constantly being conducted. This approach is considered as the most promising one for creation of new anti-aggregatory, anti-anginal, and anti-ischemic drugs based on NO-donors.
Earlier we were the first to study a new class of heterocyclic compounds--derivatives of 1,2-diazetine-1,2-dioxide--capable of nonenzymatically generating NO [10, 11]. In the course of these studies it was established that a considerable number of compounds of this series demonstrated a clearly-defined correlation between their ability to generate NO, activate soluble guanylate cyclase [12], inhibit ADP-induced human platelet aggregation, and accelerate platelet disaggregation [13].
Described in the literature there are various bicyclic N,N´-dioxides, possessing biological activity: 1,2,5-oxadiazolo[3,4-d]pyridazine-5,6-dioxides [14], substituted quinoxaline-1,4-dioxides [15] and 1,2,4-benzotriazine-1,4-dioxides [16]. In particular, we have shown that 1,2,5-oxadiazolo[3,4-d]pyridazine-5,6-dioxide is able to activate soluble guanylate cyclase and to inhibit ADP-induced platelet aggregation [14].
In the present study, the following nitro-substituted benzotetrazine-1,3-dioxides were examined: 5-nitro- (5-NBTDO), 7-nitro- (7-NBTDO), and 5,7-dinitrobenzotetrazine-1,3-dioxide (5,7-DNBTDO):
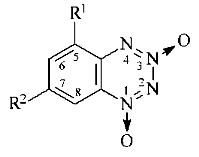
R1 = NO2, R2 = H
(5-NBTDO)
R1 = H, R2 = NO2 (7-NBTDO)
R1 = R2 = NO2 (5,7-DNBTDO)
In particular, the ability of these compounds to generate nitric oxide and activate soluble guanylate cyclase was investigated. Besides, the influence of low-molecular-weight thiols on the process of NO-formation, the enzyme activation by the benzotetrazine-1,3-dioxide derivatives, and the anti-aggregatory properties of these compounds were studied.
MATERIALS AND METHODS
Nitro-substituted benzotetrazine-1,3-dioxides (5-NBTDO, 7-NBTDO, and 5,7-DNBTDO) were synthesized according to [17]. The amount of nitric oxide formed was estimated by electron paramagnetic resonance spectroscopy (EPR spectroscopy) according to [18]. For NO-trapping, the ferrous complex of N-methyl-D-glucamine dithiocarbamate (MGD), (MGD)2-Fe2+, was used. This complex, prepared according to [19], forms the paramagnetic mononitrosyl complex with nitric oxide. In studying the role of thiols in NO generation from the compounds used, the amount of NO formed was estimated after 30 min incubation at room temperature with and without 25 µM glutathione. The molar ratio of nitrobenzotetrazine-1,3-dioxide/thiol was 1 : 5. The incubation was carried out in 15 mM HEPES buffer (pH 7.4) with dimethylsulfoxide (DMSO). The influence of compounds on guanylate cyclase activity was investigated using human platelets as a source of soluble guanylate cyclase. Platelets were isolated from blood of donors according to [20]. A suspension of washed platelets in 50 mM Tris-HCl buffer (pH 7.6) containing 0.2 mM dithiothreitol was sonicated using an MSE 5-78 ultrasonic disintegrator (UK) for 20 sec at 2°C and centrifuged for 1 h at 105,000g. The supernatant was used as the soluble guanylate cyclase preparation. Guanylate cyclase activity was assayed as described in [21]. Briefly, the samples (final volume 150 µl) contained 50 mM Tris-HCl buffer (pH 7.6), 1 mM GTP, 4 mM MgCl2, 4 mM creatine phosphate, 20 µg (120-160 units) creatine phosphokinase, 10 mM theophylline, the 105,000g supernatant (15-20 µg protein), and other additives as indicated. The effect of nitro-derivatives of benzotetrazine-1,3-dioxide on guanylate cyclase activity was studied using 10 µM concentration. Due to poor solubility of the compounds, they were initially dissolved in DMSO with subsequent dilution with 50 mM Tris-HCl buffer (pH 7.6) to the required concentration. The presence of DMSO in 10 µM nitrobenzotetrazine-1,3-dioxides had no influence on the basal activity of guanylate cyclase. The amount of cGMP formed during the enzyme reaction (15 min at 37°C) was estimated by ELISA method using kits for quantitative determination of cGMP produced by Bioimmunogen (Moscow, Russia). Protein was determined by the Bradford method [22].
In studying the effect of nitro-derivatives of benzotetrazine-1,3-dioxide on platelet aggregation, venous blood of healthy donors was collected in plastic bottles containing 1 : 10 volume of 3.8% trisodium citrate and centrifuged at 450g for 10 min at room temperature to obtain platelet rich plasma (PRP). The platelet count in PRP was adjusted with platelet poor plasma (PPP) to 2.5*108 platelets per ml. PPP was obtained by centrifugation of PRP for 30 min at 650g. Aggregation was induced by ADP in final concentration 20-40 µM causing 45-50% aggregation. Aggregation was measured according to the Born method [23] using an aggregometer designed in the Laboratory of Bioorganic Chemistry of Moscow State University, at stirring speed of 1000 rpm at 37°C. Nitrobenzo-1,3-dioxides were added before ADP.
The following reagents were used: GTP sodium salt (Fluka, Switzerland); 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide potassium salt (Carboxy-PTIO) (ICN, USA); 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) and other reagents from Sigma (USA).
RESULTS
Determination of the amount of NO formed from nitrobenzotetrazine-1,3-dioxides has shown that in the absence of thiols these compounds are unable to generate NO. In the presence of thiols (glutathione) the amount of NO formed from 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides was 1.0 ± 0.2, 5.8 ± 1.4, and 1.4 ± 0.3% of the theoretically possible amount, respectively. The results show that the compounds studied are thiol-dependent NO-donors.
Investigation of the influence of nitrobenzotetrazine-1,3-dioxides in the concentration range from 0.1 to 10 µM on the activity of soluble guanylate cyclase revealed that the maximal stimulatory effect was at their final concentration of 10 µM. As seen from Table 1, 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides, at a concentration of 10 µM, increased the enzyme activity 4.5-, 15.6-, and 8.2-fold, respectively. Note that the 105,000g supernatant which was used as an enzyme preparation contained 0.2 mM dithiothreitol (see “Materials and Methods”) whose final concentration in the sample was ~20 µM. Addition to the sample of glutathione (100 µM) increased more than twice the degree of guanylate cyclase activation by the studied compounds; at the same time, the basal activity of the enzyme remained unchanged (Table 1).
Table 1. Effect of glutathione
(100 µM) on activation of human platelet soluble guanylate
cyclase (sGC) by 10 µM 5-nitrobenzotetrazine-1,3-dioxide
(5-NBTDO), 10 µM 7-nitrobenzotetrazine-1,3-dioxide
(7-NBTDO), and 10 µM 5,7-dinitrobenzotetrazine-1,3-dioxide
(5,7-DNBTDO)
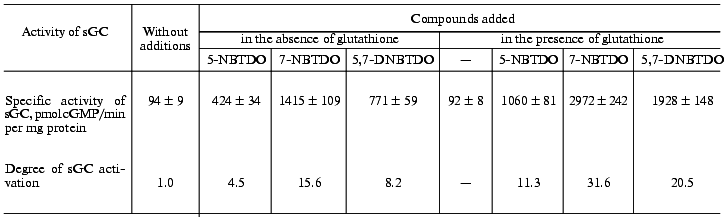
Note: The data are mean ± SD of 4-5 independent experiments.
If the stimulatory effect of benzotetrazine-1,3-dioxide derivatives is due to their ability to generate nitric oxide, it is reasonable to expect that in the presence of an NO scavenger the intensity of this effect would be lowered. Used as such NO scavenger was the potassium salt of 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (Carboxy-PTIO) [24]. Table 2 presents data on the influence of Carboxy-PTIO (50 µM) on the activation of soluble guanylate cyclase by 7-nitrobenzotetrazine-1,3-dioxide (10 µM) in the presence and absence of reduced glutathione (100 µM). As follows from these data, on addition to the sample of glutathione the degree of activation increased from 16.0- (without glutathione) to 40-fold. Simultaneously in the presence of Carboxy-PTIO (50 µM) the inhibition of the enzyme activation by 7-nitrobenzotetrazine-1,3-dioxide was increased from 15% (without glutathione) to 78% in the presence of 100 µM glutathione.
Table 2. Effect of 50 µM
2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide
(Carboxy-PTIO) on activation of human platelet soluble guanylate
cyclase (sGC) by 10 µM 7-nitrobenzotetrazine-1,3-dioxide
(7-NBTDO)
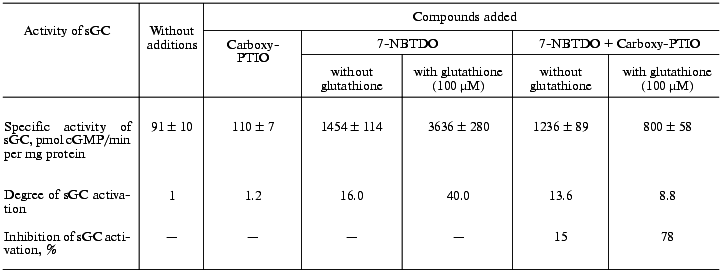
Note: The data are mean ± SD of 3-4 independent experiments.
As indicated above, the mechanism of activation of soluble guanylate cyclase by NO-donors is associated with the interaction of NO formed with the guanylate cyclase heme iron and with the formation of the nitrosyl-heme complex [4]. It is known also, that ODQ, presently used as an effective inhibitor of the heme-dependent activation of soluble guanylate cyclase can oxidize Fe2+ in the heme to Fe3+, thereby preventing NO binding and the enzyme activation [25, 26]. Table 3 shows that ODQ (in final concentration of 0.3 µM, i.e., under conditions which were earlier used by Moncada et al. [27] in analogous experiments with other NO-donors) inhibits guanylate cyclase activation by 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides by 34, 69, and 39%, respectively. These data confirm the participation of the guanylate cyclase heme in the activation of the enzyme by the compounds studied.
Table 3. Effect of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) on
activation of human platelet soluble guanylate cyclase (sGC) by
5-nitrobenzotetrazine-1,3-dioxide (5-NBTDO),
7-nitrobenzotetrazine-1,3-dioxide (7-NBTDO), and
5,7-dinitrobenzotetrazine-1,3-dioxide (7-DNBTDO) in the absence (.) and
presence of ODQ (0.3 µM) (..)

Note: The data are mean ± SD of 4-5 independent experiments.
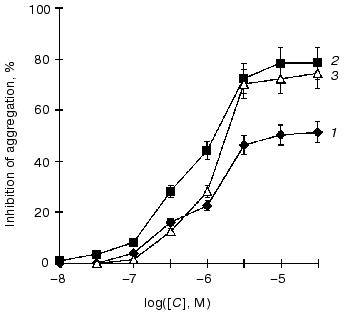
It was shown that the anti-aggregatory properties of endogenous NO and its donors are associated, to a considerable degree, with the activation of soluble guanylate cyclase and cGMP elevation in platelets [28]. We studied the ability of nitro-derivatives of benzotetrazine-1,3-dioxide to inhibit ADP-induced human platelet aggregation in vitro. As seen from the figure, the concentrations of 5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides producing 50% inhibition of platelet aggregation are estimated as 10.0, 1.3, and 2.0 µM, respectively. For comparison, the appropriate IC50 value for sodium nitroprusside is 50 µM.
Concentration-dependent inhibition of 50% irreversible ADP-induced human platelet aggregation by 5-nitrobenzotetrazine-1,3-dioxide (5-NBTDO) (1), 7-nitrobenzotetrazine-1,3-dioxide (7-NBTDO) (2), and 5,7-dinitrobenzotetrazine-1,3-dioxide (5,7-DNBTDO) (3)
DISCUSSION
The data presented here demonstrate for the first time that the compounds of a new class of bicyclic 1,3-di-N,N´-oxides (5-nitro-, 7-nitro-, and 5,7-dinitrobenzotetrazine-1,3-dioxides) are thiol-dependent donors of NO, unable to produce NO in the absence of thiols. The most effective NO-donor in this series was 7-nitrobenzotetrazine-1,3-dioxide, while 5,7-dinitro- and 5-nitrobenzotetrazine-1,3-dioxides were less effective under the same conditions. The chemical mechanism of nitric oxide formation from the above compounds in the presence of thiols has not yet been clarified. The results of preliminary experiments (A. M. Churakov and M. O. Ratnikov, unpublished data) have shown that reduction of 7-nitrobenzotetrazine-1,3-dioxide by sodium dithionate leads to formation of 5-nitrobenzotriazole. It is very likely that the reaction proceeds with formation of an intermediate compound, the appropriate N-nitrosotriazole. It is to be noted that the nitro-group of a benzene ring is not affected upon reduction. Apparently the interaction of nitro-substituted benzotetrazine-1,3-dioxides with thiols is likewise accompanied by their reduction to the respective N-nitrosobenzotriazoles, which in fact appears to be nitric oxide donors.
Nitric oxide generation predetermined the ability of nitro-substituted benzotetrazine-1,3-dioxides to activate soluble guanylate cyclase. The comparatively low degree of the enzyme activation by these compounds in the presence in sample of 20 µM dithiothreitol (Table 1) was significantly increased (by over twofold) upon addition of 100 µM reduced glutathione (Table 1), confirming the thiol-dependent mechanism of guanylate cyclase activation. 7-Nitrobenzotetrazine-1,3-dioxide proved to be the most potent enzyme activator and accordingly, the most effective nitric oxide donor in this group. The intensity of the stimulatory effects of 5,7-dinitro- and 5-nitrobenzotetrazine-1,3-dioxides was markedly lower (Table 1), consistent with the amount of NO formed from these compounds. The NO-mediated mechanism of guanylate cyclase activation was confirmed by the lowering of the stimulatory effect of 7-nitro-derivative in the presence of the standard NO scavenger, Carboxy-PTIO. Its inhibitory effect reached 78% (Table 2).
The NO- and heme-dependent mechanism of guanylate cyclase activation by benzotetrazine-1,3-dioxide derivatives was also confirmed by their lowered stimulatory effect in the presence of ODQ (Table 3) with the inhibitory effect being more pronounced in the case of 7-nitro-derivative (69%) than for 5,7-dinitro- and 5-nitro-derivatives (39 and 34%, respectively). This is apparently associated with the ability of 7-nitro-derivative to generate NO more effectively and to activate the enzyme more intensively in a heme-dependent manner. In the experiments described here, the possibility of ODQ binding to hemoglobin was entirely excluded: the enzyme preparation was obtained from highly purified platelets, which were pre-purified by use of Ficoll-Paque and did not contain any erythrocyte admixture [20].
The ability of the nitrobenzotetrazine-1,3-dioxides to activate guanylate cyclase ensures their inhibitory action on ADP-induced platelet aggregation (figure). The extent of their inhibitory effects is in full accordance with the amount of NO formed from these compounds and with the degree of guanylate cyclase activation. The most effective NO-donor and guanylate cyclase activator (7-nitrobenzotetrazine-1,3-dioxide) appears at the same time the most potent inhibitor of ADP-induced platelet aggregation (figure).
Thus, the present study has revealed a new class of thiol-dependent nitric oxide donors. The compounds of this class are able to heme-dependently activate soluble guanylate cyclase and to effectively inhibit platelet aggregation.
These studies were supported by the Russian Foundation for Basic Research (grant No. 99-04-48659).
REFERENCES
1.Kerwin, J. F., Jr., Lancaster, J. R., Jr., and
Feldman, P. L. (1995) J. Med. Chem.,38, 4343-4362.
2.Palmer, R. M. J., Ashton, D. S., and Moncada, S.
(1988) Nature,333, 664-666.
3.Murad, F. (1994) Adv. Pharmacol., 26,
19-35.
4.Craven, P., and De-Rubertis, F. (1983) Biochim.
Biophys. Acta,745, 310-321.
5.Massberg, S., Sausbier, M., Klatt, P., Bauer, M.,
Pfeifer, A., Siess, W., Fassler, R., Ruth, P., Krombach, F., and
Hoffmann, F. (1999) J. Exp. Med.,189,1255-1264.
6.Waldmann, R., and Walter, U. (1989) Eur. J.
Pharmacol., 159, 317-320.
7.Walter, U., Eigenthaler, M., Geiger, J., and
Reinhard, M. (1993) Adv. Exp. Med. Biol.,344,
237-249.
8.El-Daher, S. S., Eigenthaler, M., Walter, U.,
Furuichi, T., Miyawaki, A., Mikoshiba, K., Kakkar, V. V., and Authi, K.
S. (1996) Thromb. Haemost.,76, 1063-1071.
9.Bassenge, E., Fink, N., Skatchkov, M., and Fink, B.
(1998) J. Clin. Invest.,102, 67-71.
10.Volodarsky, L. B., and Tikhonova, L. A. (1985)
Khim. Geterotsikl. Soedin., No. 6, 748-752.
11.Kirilyuk, I. A., Utepbergenov, D. I., Mazhukin,
D. G., Fechner, K., Mertsch, K., Khramtsov, V. V., Blasg, I. E., and
Haseloff, R. F. (1998) J. Med. Chem.,41, 1027-1033.
12.Ryaposova, I. K., Grigoryev, N. B., and Severina,
I. S. (1994) Biochemistry (Moscow),59, 389-392.
13.Severina, I. S., Belushkina, N. N., and
Grigoryev, N. B. (1994) Biochem. Mol. Biol. Int., 33,
957-967.
14.Kotz, A. Ya., Grafov, M. A., Khropov, Y. V.,
Betin, V. L., Belushkina, N. N., Bussygina, O. G., Yazykova, M. Y.,
Ovchinnikov, I. V., Kulikov, A. S., Makhova, N. N., Medvedeva, N. A.,
Bulargina, T. V., and Severina, I. S. (2000) Br. J.
Pharmacol.,129, 1163-1177.
15.U. K. Patent No. 2297089 (1996).
16.Zeman, E. M., Baker, M. A., Lemmon, M. J.,
Pearson, C. I., Adams, J. A., Brown, J. M., Lee, W. W., and Tracy, M.
(1989) Int. J. Radiat. Oncol. Biol. Phys., 16,
977-981.
17.Churakov, A. M., Ioffe, S. L., and Tartakovsky,
V. A. (1991) Mend. Commun.,101-103.
18.Kurbanov, I. S., Mordvinzev, P. I., Aliev, D. I.,
and Vanin, A. F. (1989) Vopr. Med. Khim., 35, 87-91.
19.Mikoyan, V. D., Kubrina, L. N., Serezhenkov, V.
A., Stukan, R. A., and Vanin, A. F. (1997) Biochim. Biophys.
Acta, 1336, 225-234.
20.Chirkov, Yu. Yu., Tyshchuk, I. A., Belushkina, N.
N., and Severina, I. S. (1987) Biokhimiya, 52,
956-963.
21.Garbers, D. L., and Murad, F. (1979) Adv.
Cycl. Nucl. Res.,10, 57-67.
22.Bradford, H. M. (1976) Analyt.
Biochem.,72, 248-254.
23.Born, G. V. R. (1962) Nature, 194,
927-929.
24.Akaike, T., Yoshida, M., Miyamoto, Y., Sato, K.,
Kohno, M., Sasamoto, K., Miyzaki, K., Ueda, S., and Maeda, H. (1993)
Biochemistry,32, 827-832.
25.Garthwaite, J., Southam, E., Boulton, C. L.,
Schmidt, K., and Mayer, B. (1995) Mol. Pharmacol., 48,
184-198.
26.Zhao, Y., Brandish, P. E., Divalentin, M.,
Schelvis, J. P. M., Babcook, G. T., and Marletta, M. A. (2000)
Biochemistry, 39, 10848-10854.
27.Moro, M. A., Rassel, R. J., Cellek, S.,
Lizasoain, I., Su, Y., Darley-Usmar, V. M., Radomski, M. W., and
Moncada, S. (1996) Proc. Natl. Acad. Sci. USA,93,
1480-1485.
28.Mellion, B. T., Ignarro, L. J., Ohlstein, E. H.,
Pontecorvo, E. G., Hyman, A. L., and Kadowitz, P. J. (1981)
Blood,57, 946-955.