REVIEW: The Fibrinolysis System: Regulation of Activity and Physiologic Functions of Its Main Components
A. B. Dobrovolsky* and E. V. Titaeva
Russian Cardiology Research Center, 3-ya Cherepkovskaya ul. 15a, Moscow, 121552 Russia; fax: (095) 140-9839; E-mail: cclibr@transts.ru* To whom correspondence should be addressed.
Received June 14, 2001; Revision received August 9, 2001
This review summarizes basic properties and mechanisms of activation and inhibition of main components of the fibrinolytic system that, acting in accord with the system of blood coagulation, provides temporal formation of fibrin clots at sites of vascular injury for the time required for the regeneration of vascular wall. Impairments in the fibrinolytic system may cause bleedings or thrombotic complications in patients. The predictive value of some components of the fibrinolytic system with respect to the development of complications of atherothrombosis is considered.
KEY WORDS: plasminogen activators, inhibitors of fibrinolysis, thrombosis, risk factors, cardiovascular diseases
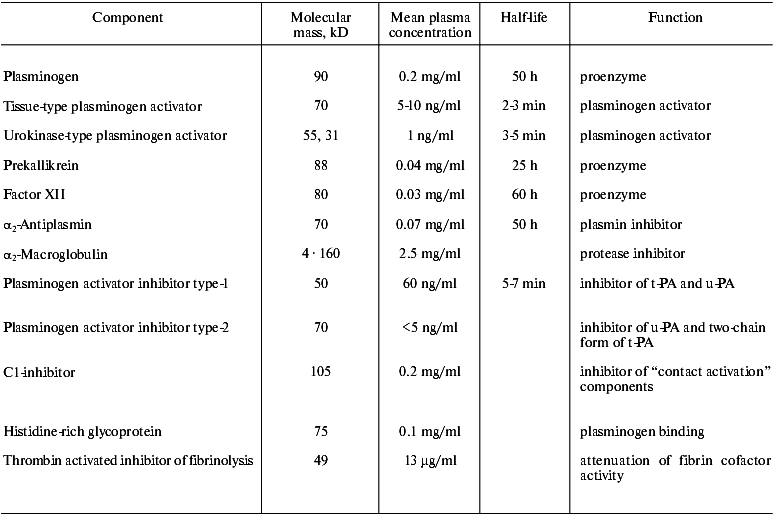
Abbreviations: alpha2-AP) alpha2-antiplasmin; alpha2-MG) alpha2-macroglobulin; PAI-1) type-1 inhibitor of plasminogen activators; TAFI) thrombin activated inhibitor of fibrinolysis; t-PA) tissue type plasminogen activator; u-PA) urokinase type plasminogen activator.
Until the 1990s investigations of the fibrinolytic system were focused
mainly on the study of the mechanisms of activation of plasminogen (Pg)
and plasmin-mediated degradation of intravascular fibrin depositions,
probably because for a long time it was well-known that impairments in
this system are associated with bleedings or thrombotic complications
in patients. However, in the last decade, when methods of gene
inactivation in mice were applied for the study of the physiological
role of proteins, it became evident that functions of components of the
fibrinolytic system are not restricted to the dissolution of fibrin.
Now the participation of this system in the regulation of cellular
activity and tissue development is under intensive exploration.
It should be mentioned that although significant progress has been achieved in the study of the structure-function relationships and mechanisms of activation and inhibition of the main components of fibrinolytic system, nevertheless some details of the regulation of fibrinolysis in vivo remain to be elucidated. For example, fibrin is a pathologic formation yet a structure that protects from bleeding at the site of vascular injury. Its removal is necessary for the restoration of normal blood flow, but this should occur only after the regeneration of the vessel wall. At the same time, tissue-type plasminogen activator (t-PA), which is thought to be the primary initiator of fibrinolysis in the circulation, is the only protease of the hemostatic system that is continuously secreted by the endothelium in active form. What does postpone the lysis of fibrin for the time needed for the regeneration of vessel wall? In the absence of fibrin, t-PA activates plasminogen at a very low rate, and because of high activity of plasminogen activator inhibitor type-1 (PAI-1) in blood t-PA is inactivated with half-time of about 2 min. It thus appears that most of the secreted t-PA will be inactivated by PAI-1 before it can bind with a fibrin clot. From this point of view, t-PA and PAI-1 represent a “suicide pair”, or one may assume that these components and/or “inactive t-PA-PAI-1 complex” have some other function(s) besides the regulation of fibrinolysis [1]. Obviously, further studies are needed for the elucidation of mechanisms that determine the stability of thrombi in vivo. Impairment of these mechanisms may predispose to bleeding or thrombosis in patients.
MAIN COMPONENTS OF THE FIBRINOLYTIC SYSTEM
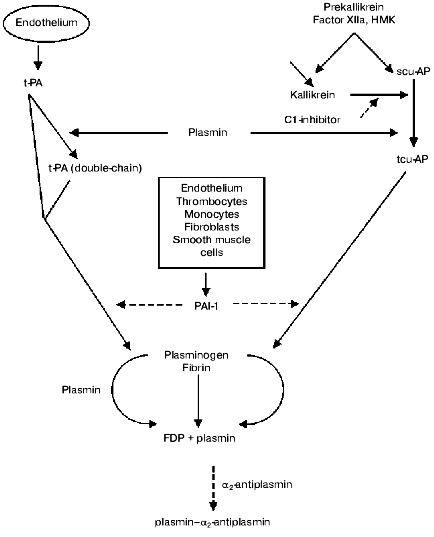
The fibrinolytic system comprises of a proenzyme (plasminogen), enzymes that proteolytically activate plasminogen, and several inhibitors that regulate activation of plasminogen, activity of plasmin, and stepwise degradation of fibrin (Fig. 1, table).
Main components of the fibrinolytic systemFig. 1. The fibrinolytic system. Solid arrows represent secretion and activation of the components, dotted arrows their inhibition. t-PA activates with high rate only fibrin bound plasminogen. tcu-AP can activate both free and fibrin bound plasminogen. Plasmin released from a thrombus with fibrin degradation products is rapidly inactivated by alpha2-antiplasmin (FDP is a product of fibrinogen degradation).
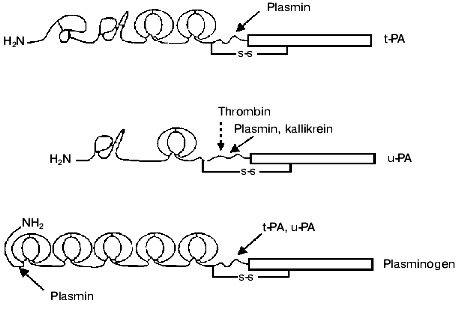
Plasminogen activators. Tissue-type plasminogen activator (t-PA) is synthesized and secreted by endothelial cells as a single chain active enzyme with Mr of 70,000. t-PA is composed of five domains: the N-terminal domain homologous to the finger-like domain of fibronectin is followed by a domain homologous to the epidermal growth factor domain (EGF), two kringle-like domains of plasminogen, and C-terminal domain homologous to trypsin-like proteases (Fig. 2). The active site is composed of His322, Asp371, and Ser478 [2]. The finger-like and kringle-2 domains possess two distinct fibrin-binding sites. The binding of t-PA with PAI-1 involves the interactions with the active site as well as with regions of residues 296-304 and kringle-2 domain [3]. Inactive complex t-PA-PAI-1 retains some affinity to fibrin and therefore it can compete with free t-PA in the binding with fibrin. The application of recombinant technology has allowed large-scale production of t-PA and its use for thrombolytic therapy. Wide use of recombinant t-PA for thrombolysis in patients with myocardial infarction stimulated intensive studies of structure-function relationships of t-PA and development of mutant forms of t-PA with improved pharmacokinetic properties, i.e., with prolonged lifetime in the circulation and increased resistance to inhibitors and to cleavage by plasmin (reviewed in [4, 5]).
Plasmin, kallikrein, and factor Xa can split the Arg275-Ile276 peptide bond with formation of a two-chain form of t-PA. The splitting increases activities towards synthetic chromogenic substrates and inhibitors and the rate of activation of plasminogen in the absence of fibrin, but in the presence of fibrin both forms of t-PA activate plasminogen with similar rates [6].Fig. 2. Domain structure of t-PA, u-PA, and plasminogen. t-PA consists of the finger-like domain of fibronectin, epidermal growth factor domain, two kringles, and the catalytic domain. The binding of t-PA to fibrin is mediated by the finger-like and kringle-2 domains, and binding to PAI-1 is mediated by the kringle-2 domain and the residues 296-304 of the N-terminal region of the catalytic domain. scu-PA consists of the epidermal growth factor domain and the kringle and catalytic domain. Plasmin and kallikrein splits scu-PA at Lys158 with formation of fully active two-chain enzyme. Thrombin can split scu-PA at Arg156 with formation of a two-chain derivative that does not activate free plasminogen, but can be slowly activated after splitting by plasmin at Lys158. Plasminogen consists of five kringle domains, mediating binding with fibrin, alpha2-antiplasmin, and cell receptors and catalytic domain. Plasminogen activation occurs after splitting at Arg561. Plasmin can cleave peptide bonds after Arg67, or Lys76,77 producing truncated forms of plasminogen which have higher affinity for fibrin and are activated by t-PA or u-PA more rapidly than the parent molecule.
In the absence of fibrin, t-PA activates plasminogen with very low rate. The reaction is characterized by a low rate constant of about 0.01-0.06 sec-1 and Km´ of about 60 µM, which is 30 times higher than the concentration of plasminogen in blood. When fibrin is present, both t-PA and plasminogen bind to fibrin. The formation of ternary complex increases the catalytic efficiency of t-PA by 1000-fold [7]. The activation of plasminogen by t-PA can be stimulated not only by fibrin. It has been shown that cell membranes and proteins of extracellular matrix, such as thrombospondin and collagen IV, increase the rate of plasminogen activation, but their “cofactor” efficiency is significantly less than that of fibrin [8].
Urokinase. This trypsin-like protease with the ability to activate plasminogen was isolated first from urine and was thus named urokinase. The isolated enzyme consisted of two polypeptide chains linked by a disulfide bond. Later it was shown that many cell types synthesize and secrete urokinase as a single chain 54-kD glycoprotein, which does not hydrolyze synthetic chromogenic substrates and does not react with acylating reagents. This form of the activator was named prourokinase, but further studies revealed that the single chain form of urokinase is not a true proenzyme and it was recommended to designate two forms of enzyme as single-chain urokinase-type plasminogen activator (scu-PA), and two-chain urokinase-type plasminogen activator (tcu-PA). The scu-PA molecule is composed of three domains: N-terminal domain homologous to epidermal growth factor domain (EGF) is followed by domain homologous to the kringle-like domains of plasminogen, and C-terminal domain homologous to trypsin-like proteases (Fig. 2). The active site is formed by His204, Asp255, and Ser356.
The scu-PA molecule can be cleaved specifically at several sites. Plasmin or kallikrein cleaves the Lys158-Ile159 peptide bond producing fully active urokinase consisting of two polypeptide chains connected by a disulfide bond. Plasmin can cleave further the Lys135-Lys136 peptide bond in tcu-PA with the liberation of N-terminal fragment containing regulatory domains and formation of a 33-kD variant of tcu-PA. Stromelysin-1 cleaves the Glu143-Leu144 bond with formation of a 32-kD variant of scu-PA. Thrombin cleaves the Arg156-Phe157 peptide bond in scu-PA with formation of a variant of tcu-PA that does not activate plasminogen in purified systems, but can induce thrombolysis in vivo. The Lys158-Ile159 peptide bond in thrombin-cleaved tcu-PA can be hydrolyzed by plasmin with formation of active enzyme (reviewed in [4, 5]).
scu-PA does not bind specifically with fibrin; nevertheless, intravenous infusion of scu-PA induces lysis of thrombi without significant activation of plasminogen in the circulation. It was suggested, that the clot-specific action of scu-PA could be due to: 1) formation in blood of a complex with reversible inhibitor, which dissociates in the presence of fibrin; 2) local activation of scu-PA at the surface of thrombus by kallikrein, factor XIIa, or plasmin; 3) inclusion into a clot of different proteins and cells that may form a unique structure responsible for local activation of plasminogen [4, 5].
Many cell types express the specific receptors for u-PA (u-PAR). u-PAR is a cysteine-rich 65-kD glycoprotein comprised of three homologous extracellular domains. u-PAR is anchored to the cell surface by a covalent bond between the C-terminus of the receptor and membrane glycosylphosphatidylinositol. Binding of u-PA with u-PAR involves the interaction between the EGF-domain of the enzyme and the N-terminal domain of the receptor and is characterized by high affinity (Kd ~10-9-10-10 M) and specificity, since other EGF-containing proteins do not interact with u-PAR. Binding of u-PA to u-PAR activates local proteolysis and intracellular signal transduction, processes which play a critical role in tissue development (reviewed in [9-11]).
The presence of two different physiological activators of plasminogen raises the question about their relative roles in homeostasis. Studies of the viability and growth of mice with individual and combined deficits of u-PAR, u-PA, or t-PA provided evidence that u-PA and t-PA are complementary activators of plasminogen with a high functional overlap. Taking into account the difference of functional properties of u-PA and t-PA, Bugge et al. [12] suggested that u-PA can be of primary importance for cell-mediated activation of plasminogen in tissues, whereas t-PA, possessing a high affinity for fibrin, can be of primary importance for the lysis of fibrin clots in the circulation.
Inhibitors of plasminogen activators. Four different proteins belonging to the superfamily of serine protease inhibitors (serpins) have been identified as plasminogen activator inhibitors. In normal blood, t-PA and tcu-PA are inhibited mainly by a 52-kD glycoprotein that is synthesized by the endothelium and is named plasminogen activator inhibitor type-1 (PAI-1). Plasminogen activator inhibitor type-2 (PAI-2) inactivating with high rate tcu-PA is synthesized by the placenta, monocytes, and macrophages. In normal blood PAI-2 is not detected, but it may appear in various diseases. Inhibitors described as PAI-3 and PAI-4 were later identified as an inhibitor of protein C and the protease nexin I, respectively (reviewed in [13, 14]). The inhibition of plasminogen activators by PAI-1 proceeds in two steps. In the first step a reversible complex is formed with a high rate (kcat ~ 107 M-1*sec-1), then the inhibitor is slowly cleaved at the reactive site Arg346-Met347. The C-terminal peptide of the inhibitor is released, but the carboxyl group of Arg346 remains covalently bound with Ser of the active site of the activator [15].
A large amount of PAI-1 (~90% of the total content in blood) is stored in the alpha-granules of platelets. Upon activation, platelets release PAI-1, significantly increasing the local concentration of PAI-1 at sites of thrombus formation, and thus platelets may increase the proteolytic stability of fibrin matrix [16].
In plasma and other biological fluids, PAI-1 is present in two forms, active and latent, differing in three-dimensional structure. In the active form the reactive center of PAI-1 is exposed on the surface, while in the latent form it is immersed into the protein globule. In plasma the active form of PAI-1 is spontaneously transformed into the latent form that can be reactivated in vitro by treatment with denaturing agents. The physiological role and mechanism of in vivo activation of latent PAI-1 is not known. However, it should be mentioned that Lambers et al. [17] showed the activation of latent PAI-1 by phospholipid vesicles containing phosphatidylserine or phosphatidylinositol. This indicates that at sites of vessel injury activated platelets or membranes of disrupted cells may activate latent PAI-1 and thus increase the proteolytic stability of a thrombus. Active PAI-1 interacts specifically with vitronectin. The formation of the complex alters the properties of both proteins. It stabilizes the active conformation of PAI-1 and decreases binding of vitronectin to cellular receptors [1].
The synthesis of PAI-1 is affected by a large number of substances: insulin, cytokines, transforming growth factor beta, thrombin, atherogenic lipoproteins, and others. The level of PAI-1 is rapidly increased several fold during acute phase of disease. Therefore, the concentration and activity of PAI-1 in plasma of healthy individuals and in patients with various diseases vary greatly. Additionally, the level of PAI-1 is subject to circadian variations: seasonal with highest levels in the winter and lowest in the summer, and diurnal with highest levels early in the morning and lowest in the evening. Morning elevation of PAI-1 precedes for a few hours the peak of onset of acute myocardial infarction, sudden cardiac death, and ischemic stroke, caused mainly by thrombotic occlusion of ruptured atheromatous plaque. Variability of PAI-1 level in blood is higher than that of other components of the fibrinolytic system. This indicates that PAI-1 may be considered as a main determinant of fibrinolytic activity in vivo. Direct evidence for the relationship between level of PAI-1 expression and thrombosis was obtained in mice transgenic for human PAI-1 cDNA attached to the promoter, which provided temporal expression of this gene. In these animals, venous thrombi were developed during the period of enhanced synthesis of PAI-1 and resolved after decline of PAI-1 expression [18].
Plasminogen. Plasminogen is secreted by the liver as a single chain 92-kD glycoprotein consisting of 791 amino acid residues with Glu as the N-terminal amino acid. The molecule is composed of five kringle-like domains containing “lysine-binding sites” and C-terminal domain homologous to other trypsin-like proteases (Fig. 2). The kringle-1 domain binds lysine with high affinity (Kd ~ 9 µM), other domains bind lysine with low affinity (Kd ~ 5 mM). The kringle domains mediate interactions of plasminogen and plasmin with substrates, inhibitors, and cell membranes, which are crucial for the activation of plasminogen and localization of proteolytic activity of plasmin. Plasminogen can be cleaved by plasmin at peptide bonds between Arg67-Met68, Lys76-Lys77, or Lys77-Val78 with the release of an 8-kD N-terminal polypeptide and formation of Lys-plasminogen, which in the absence of fibrin is activated with higher rate than Glu-plasminogen.
The splitting of the Arg561-Val562 peptide bond converts plasminogen to the active enzyme plasmin consisting of two polypeptide chains linked by a disulfide bond. The N-terminal heavy A-chain possesses all the kringle domains. The C-terminal light B-chain contains the catalytic site composed of His602, Asp645, and Ser740. The light chain isolated from plasmin after the reduction of the disulfide bond displays substrate specificity similar to that of trypsin. The specificity of plasmin is provided by domains of its heavy chain (reviewed in [19, 20]).
Inhibitors of plasmin. alpha2-Antiplasmin (alpha2-AP) is a member of the serpin superfamily of inhibitors. alpha2-AP is secreted by the liver as a single chain 70-kD glycoprotein consisting of 464 amino acid residues. In the blood, alpha2-AP exists in the two molecular forms: Met-alpha2-AP and Asn-alpha2-AP. Asn-alpha2-AP is formed after the splitting off a 12-amino acid peptide from the N-terminal end of Met-alpha2-AP. Both forms of alpha2-AP inhibit plasmin with similar rates, but Met-alpha2-AP has a lower affinity for fibrin than Asn-alpha2-AP [21].
When plasmin is added to blood plasma it is rapidly inactivated by alpha2-AP (t1/2 ~ 0.1-0.5 sec). The inactivation proceeds in two steps: very rapid formation of a stoichiometric reversible complex (Kd ~ 2*10-10 M) followed by slower cleavage of the Arg364-Met365 peptide bond at the reactive center of alpha2-AP with formation of covalent bond between Ser of the active site of plasmin and the carboxyl group of Arg364 of the N-terminal part of alpha2-AP. The C-terminal fragment of alpha2-AP remains associated noncovalently with the kringle-1 domain of plasmin. The strong and specific interaction of alpha2-AP with a substrate-binding site of plasmin provides the fibrin specificity of this enzyme. In the presence of fast-acting inhibitor, plasmin retains its activity until it is bound to fibrin [22].
alpha2-AP plays an important role in the regulation of lysis of fibrin matrix of a thrombus. When fibrin is formed, it binds plasminogen and alpha2-AP in approximately equimolar quantities. It is noteworthy that alpha2-AP is bound and then cross-linked by factor XIIIa to the C-terminal part of the alpha-chain of fibrinogen, which is degraded by plasmin first [22].
Fibrinogen has symmetrical structure formed by three pairs of non-identical polypeptide chains designated as Aalpha-, Bbeta-, and gamma-chains. Fibrin(ogen) is sequentially digested by plasmin with formation of degradation products designated as X-, Y-, D-, and E-fragments. The sites most sensitive to proteolysis are Lys/Arg at positions 584, 425, and 207 of the alpha-chain and 42 of the beta-chain of fibrinogen. Cleavage at these sites may proceed asymmetrically in both halves of the fibrinogen molecule resulting in a set of X-fragments with molecular masses ranged from 330 to 240 kD. X-fragments possess central and peripheral sites of polymerization; therefore, after these initial cleavages fibrin still retains polymer structure. Degradation of fibrin clots to soluble fragments (Y-, D-, and E-fragments) occurs after the splitting off from fibrin monomers of at least one D-fragment [23].
It seems likely that physiological role of alpha2-AP cross-linked to fibrin consists in inactivation of plasmin, generating from plasminogen trapped during formation of fibrin clot, after initial cleavages, which do not destroy the fibrin network, but open new high affinity binding sites for plasminogen and t-PA. Final degradation of the fibrin network is done by plasmin, generating from plasminogen adsorbed from blood by partially degraded fibrin. Indeed, Sakharov et al. [24] have shown superficial accumulation of plasminogen and layer-by-layer reduction in size of fibrin clots during the lysis. The presence of two sequential phases in lysis of fibrin may be one of the mechanisms providing temporary stability of fibrin clots at sites of vascular injury [19].
alpha2-Macroglobulin (alpha2-MG) is a 725-kD glycoprotein composed of four identical polypeptide chains [25] containing amino acid sequence Arg681-Val-Gly-Phe-Tyr-Glu686 with polypeptide bonds that can be cleaved by different types of proteases. Cleavage induces conformational changes in alpha2-MG resulting in the hydrolysis of an intramolecular thioether bond between side chains of Cys949 and Gln952 and introduction of a new intermolecular covalent bond between the gamma-carbonyl group of Gln952 of alpha2-MG and the epsilon-NH2 group of a Lys residue of the protease. Unlike serpins, alpha2-MG does not modify the catalytic site of the protease. Therefore, the alpha2-MG-protease complex can split low molecular weight substrates, but binding and cleavage of proteins is impaired because of hindrances created by an attached inhibitor (reviewed in [19]). Inactivation of plasmin by alpha2-MG proceeds with much slower rate than that by alpha2-AP. Nevertheless, alpha2-MG can play an important role in the inactivation of plasmin after consumption of alpha2-AP that is present in plasma at a concentration two times lower than that of plasminogen.
Thrombin-activated fibrinolysis inhibitor. Investigations of mechanisms of profibrinolytic effects of activated protein C and antifibrinolytic action of thrombin have revealed a novel inhibitor of fibrinolysis named thrombin-activated fibrinolysis inhibitor (TAFI). It was soon found that TAFI is similar or identical to the earlier described carboxypeptidase U and plasma procarboxypeptidase B [26, 27]. The mechanism of antifibrinolytic action of TAFI consists in the removal of C-terminal Lys or Arg residues exposed during initial cleavages of fibrin by plasmin and forming new high affinity binding sites for plasminogen and t-PA. The removal of these binding sites abrogates the acceleration of lysis rate observed after initial cleavages of fibrin [24, 28].
TAFI is secreted by the liver as a single chain 58-kD glycoprotein, consisting of 401 amino acid residues. It is activated after the cleavage by trypsin-like proteases at Arg92 yielding N-terminal activation peptide and a 35-kD enzyme with carboxypeptidase B-like activity [29]. The physiological activators of TAFI can be thrombin or plasmin. Activation of TAFI by free thrombin proceeds with low rate due to unfavorable kinetic parameters of the reaction: kcat is about 0.002 sec-1, and Km´ ~ 2 µM, which is 10 times higher than the concentration of TAFI in plasma. Binding of thrombin to thrombomodulin increases kcat to ~1.2 sec-1 and decreases Km´ to ~1 µM resulting in 1250-fold increase of the catalytic efficacy of the reaction [30]. Plasmin also activates TAFI with low rate (kcat ~ 0.0004 sec-1, and Km´ ~ 0.055 µM). Activation is markedly stimulated by glycosaminoglycans and at therapeutic concentrations of unfractionated heparin in blood the catalytic efficacy of plasmin could reach 1/10 of that of thrombin-thrombomodulin complex [31].
A physiological inhibitor of TAFI has not been identified. In solutions, the activity of TAFI decays spontaneously. Inactivation is associated with conformational changes of TAFI. The rate of inactivation increases with increasing temperature (t1/2 is about 3 h at 22°C and ~8 min at 37°C) and decreases in the presence of competitive inhibitors (Lys and its analogs) and substrates. Thus, in blood antifibrinolytic action of TAFI is limited mainly by intrinsic instability of the free form of this enzyme [32].
The discovery of TAFI and elucidation of the mechanism of its action explained why clots formed from blood of patients with hemophilia are degraded more rapidly that those from normal individuals. The amount of thrombin formed during clotting of plasma deficient in components of the intrinsic pathway of coagulation is not sufficient for proper activation of TAFI and attenuation of fibrinolysis provided by this component in normal plasma [27, 30].
Components of the protein C system may exert opposite effects on the activation of TAFI. On one hand, binding of thrombin to thrombomodulin stimulates the activation of protein C and subsequent degradation of factors Va and VIIIa. This abrogates the generation of thrombin and should decrease the extent of TAFI activation within clots. It is well known that both deterioration of the protein C system and resistance of factor V-Leiden to cleavage by protein C predispose patients to thromboembolic diseases [33]. On the other hand, thrombomodulin stimulates activation of TAFI by thrombin to the same extent as that of protein C. Both protein C and TAFI circulate in blood at concentrations which are 10 times lower than the corresponding Km´ values for the thrombin-thrombomodulin complex. This means that the extent of activation of both proenzymes depends on concentrations of all components of the reaction. Mosnier et al. [34] studied the influence of concentration of added thrombomodulin on t-PA-stimulated lysis of clots produced from normal plasma. They showed that at low concentrations of thrombomodulin (from 0.05 to 5 nM) the activation of TAFI predominated, resulting in the prolongation of lysis time. At higher concentrations of thrombomodulin the activation of protein C predominated, leading to abrogation of thrombin generation and TAFI activation and to increase of lysis rate. Thus, these data indicate that thrombomodulin may exert anticoagulant as well as antifibrinolytic (i.e., procoagulant) actions. The balance between these activities of thrombomodulin depends on many factors determining formation of thrombi in vivo.
The mean plasma concentration of TAFI is about 200 nM [35]. Large variations (4-6-fold) of concentration of TAFI were found even among healthy individuals, and time of t-PA-induced lysis of normal plasma clots correlated positively with the content of TAFI [36, 37]. It should be mentioned that the half-maximal inhibition of fibrinolysis in vitro was observed at 1 nM TAFI and maximal at 10 nM. This means that in spite of low kcat and high Km´ values TAFI is activated during clotting of normal plasma to an extent sufficient for downregulation of fibrinolysis [32, 36, 38].
Data confirming a significant role of TAFI in the stabilization of thrombi were obtained in experiments on thrombolysis. Infusion of specific inhibitor of carboxypeptidase B from potato tubes (Ki ~ 0.4 nM) enhanced 2-4-fold the efficacy of t-PA in lysis of arterial thrombi in rabbits [39-41]. However, it should be mentioned that in vitro inhibitory effect of TAFI gradually decreased when the concentration of t-PA increased. This may be due to pronounced activation of plasminogen causing consumption of alpha2-AP, degradation of fibrinogen, and formation of Lys-plasminogen, which has higher affinity to fibrin and is activated by t-PA in the absence of fibrin at higher rate than Glu-plasminogen. This suggestion is supported by the observation that when clot lysis was stimulated by activator from the saliva of the vampire bat, possessing much higher fibrin-specificity than t-PA, an inhibitory effect of TAFI was observed at all studied concentrations of this activator [42].
The role of TAFI may not be restricted to the inhibition of fibrinolysis. By splitting off C-terminal Lys and Arg residues it may participate in the regulation of activity of different proteins and bioactive peptides [43]. Clinical study of the (patho)physiological role of TAFI are now in progress. Van Tilburg et al. [44] showed that increased levels of TAFI predispose to deep vein thrombosis. The concentration of TAFI increases with age and in women below 50 years receiving oral contraceptives or hormone replacement therapy [35, 37]. Significant decrease of TAFI was observed in patients with chronic liver disease [45] and in patients with acute promyelocytic leukemia, diseases associated with high rate of hemorrhages [46].
COMPONENTS OF THE FIBRINOLYTIC SYSTEM IN PATIENTS WITH
CARDIOVASCULAR DISEASES
Morphological and angiographic studies have demonstrated that formation of thrombi at sites of atherosclerotic lesions is the major cause of development of clinical complications of atherosclerosis, which are leading contributors to morbidity and mortality throughout the industrialized world. The thrombogenicity of atherosclerotic plaque is determined mainly by the stability of fibrous cap and contents of tissue factor in its core [47], which activates the coagulation cascade when exposed to flowing blood [48].
Thrombotic occlusion at the site of vessel injury occurs when the growth rate of thrombus exceeds that of its lysis. Indeed, several prospective studies showed that impairments of fibrinolysis predispose to complications of atherosclerosis [49-53]. PAI-1 is considered as a main determinant of fibrinolytic activity; therefore, association of increased levels of this inhibitor with thrombotic complications observed in many studies is not surprising. However, it should be mentioned that in multivariate analysis predictive value of PAI-1 is attenuated indicating association of PAI-1 level with other risk factors, such as markers of insulin resistance and hypercholesterolemia [54, 55].
Moreover, PAI-1 is an acute phase protein that may increase several-fold in response to illness. For example, in patients hospitalized with acute myocardial infarction we observed a direct relationship between PAI-1 activity and time elapsed from the symptom onset [56]. Moreover, duration of PAI-1 increase and its magnitude were higher in patients with occluded arteries than in those with coronary reperfusion within 6 h after admission. These data are consistent with those of Paganelli et al. [57]. Indirect evidence of the significance of PAI-1 in conferring proteolytic stability of thrombi may be drawn from the observation that thrombolytic efficacy of t-PA displays circadian variation opposite to that of PAI-1 activity [58]. It should be mentioned that increased stability of thrombi in the morning might also be due to the phenomenon of “morning hypercoagulability”. Enhanced generation of thrombin may result in higher extent of TAFI activation that inhibits fibrinolysis; however, this remains to be explored.
We studied relationships between markers of activation of blood coagulation and fibrinolysis in patients with various degrees of atherosclerosis who did not have clinical symptoms of acute thrombosis at admission [59, 60]. In these patient we observed increase of plasma levels of beta-thromboglobulin, fragment 1+2 of prothrombin, and D-dimer with increase of the extent of atherosclerotic lesions. D-dimer correlated positively with activity of t-PA and concentration of plasmin-antiplasmin complex and negatively with activity of PAI-1. This agrees with data of van der Bom et al. [61] that among patients with peripheral atherosclerosis high levels of D-dimer were found in those who had low PAI-1 activity. Thus, in patients with atherosclerotic lesions but without symptoms of acute thrombosis, fibrin formation may be limited to some extent by active fibrinolysis that protects from total occlusion of injured vessels. However, this state is not stable. In our prospective study of 121 patients with peripheral atherosclerosis, 32 thrombotic events (half of them with fatal outcome) were identified during five years of follow-up. Multivariate analysis revealed that high level of D-dimer in plasma is an independent risk factor of thrombotic complications in patients with atherosclerotic lesions of peripheral arteries [62].
The concentration of t-PA in plasma is much lower than that of PAI-1 and other proteases. Therefore, precise measurement of its activity in plasma is difficult. Measurement of t-PA by immunoassay revealed “paradoxical” results--association of elevated levels of t-PA antigen in plasma with the risk of cardiovascular events, positive correlation of t-PA antigen with PAI-1 activity, and negative correlation between antigen of t-PA and its activity [50, 63, 64]. This associations may be explained by data of Chandler et al. [65] that inactive t-PA-PAI-1 complex is cleared from circulation with lower rate than free t-PA. Thus, elevation of t-PA antigen in plasma may be expected when the rate of t-PA inactivation is increased. Recently Wiman et al. [66] using immunoassays discriminating between free t-PA and its complexes with inhibitors showed that more than half of total t-PA antigen in plasma is present as t-PA-PAI-1 complex, and that concentration of the former is a stronger predictor for recurrent myocardial infarction than that of total t-PA antigen.
High rate of thrombotic complications in patients with atherosclerosis determined intensive studies of methods of their prevention, which should be directed toward stabilization of atheroma (ideally its regression) and prevention of thrombi growth.
Large trials showed that the use of inhibitors of platelet aggregation (aspirin, ticlopidine) can reduce the risk of cardiovascular events by 25-30% [67, 68]. Now new antiplatelet drugs, antagonists of IIb/IIIa receptors inhibiting the final step of platelet aggregation and consequently the action of all platelet agonists, are under intensive exploration (reviewed in [69]).
Thrombin generated during activation of clotting activation participates not only in formation and stabilization of thrombi, but also in activation of different cells involved in processes of inflammation and tissue regeneration (reviewed in [70]). Therefore, the use of drugs inhibiting activity and/or generation of thrombin could influence the stability of plaques. Development of low molecular weight heparins that could be used for relatively long-term treatment of outpatients provided new opportunities for prevention of thrombotic complications in patients with atherosclerosis [71].
Large placebo-controlled trials have shown that lipid lowering therapy significantly reduces the risk of cardiovascular complications of atherosclerosis (reviewed in [72]). It is noteworthy that the most effective is treatment with statins, which lower cholesterol by inhibiting synthesis of mevalonic acid. Recent studies demonstrated that protective action of statins may consist not only in lowering of atherogenic lipoproteins, but also in regulation of the synthesis of other factors including vasoactive compounds, tissue factor, PAI-1, and matrix metalloproteinases, which may influence the stability and thrombogenicity of atheroma [73-76].
The multifactorial nature of atherosclerosis with complex relationships between participating factors suggests that for effective treatment of patients identification of predisposing factor and search for optimal combinations of drugs affecting different mechanisms of disease are needed.
REFERENCES
1.Stefansson, S., Haudenschild, C. C., and Lawrence,
D. A. (1998) Trends Cardiovasc. Med., 8, 175-180.
2.Pennica, D., Holmes, W. E., Kohr, W. J., Harkins,
R. N., Vehar, G. A., Ward, S. A., Bennett, W. F., Yelverton, E.,
Seeburg, P. H., Heyneker, H. L., Goeddel, D. V., and Collen, D. (1983)
Nature, 301, 214-221.
3.Madison, E. L., Goldsmith, E. J., Gerard, R. D.,
Gething, M. J. H., Sambrook, J. F., and Bassel-Duby, R. S. (1990)
Proc. Natl. Acad. Sci. USA, 87, 3530-3533.
4.Verstraete, M. (2001) Thrombosis,
Hemostasis and Rheology, No. 15, 4-13.
5.Maksimenko, A. V. (1995) Mol. Biol.
(Moscow), 29, 38-60.
6.Tate, K. M., Higgins, D. L., Holmes, W. E.,
Winkler, M. E., Heyneker, H. L., and Vehar, G. A. (1987)
Biochemistry, 26, 338-343.
7.Norman, B., Wallén, P., and Rånby, M.
(1985) Eur. J. Biochem., 149, 193-200.
8.Stack, S., Gronzales-Gronow, M., and Pizzo, S. V.
(1990) Biochemistry, 29, 4966-4970.
9.Chapman, H. A., Wei, Y., Simon, D. I., and Waltz,
D. A. (1999) Thromb. Haemost., 82, 291-297.
10.Blasi, F. (1999) Thromb. Haemost.,
82, 298-304.
11.Koshelnick, Y., Ehart, M., Stockinger, H., and
Binder, B. R. (1999) Thromb. Haemost., 82, 305-311.
12.Bugge, T. H., Flick, M. J., Danton, M. J. S.,
Daugherty, C. C., Rømer, J., Danø, K., Carmeliet, P., Collen,
D., and Degen, J. L. (1996) Proc. Natl. Acad. Sci. USA,
93, 5899-5904.
13.Krishnamurti, C., and Alving, B. (1992) Semin.
Thromb. Hemost., 18, 67-80.
14.Juhan-Vague, I., and Alessi, M. C. (1997)
Thromb. Haemost., 78, 656-660.
15.Thorsen, S., and Philips, M. (1984) Biochim.
Biophys. Acta, 802, 111-118.
16.Kruithof, E. R., Tran-Thang, C., and Bachman, F.
(1986) Thromb. Haemost., 55, 201-205.
17.Lambers, J. W. J., Cammenga, M., Konig, B. W.,
Mertens, K., Pannekoek, H., and van Mourik, J. A. (1987) J. Biol.
Chem., 262, 17492-17496.
18.Erickson, L. A., Fici, G. J., Lund, J. E., Boyle,
T. P., Polites, G. H., and Marotti, K. R. (1990) Nature,
346, 74-76.
19.Bachmann, F. (1987) in Thrombosis and
Haemostasis (Verstraete, M., Vermylen, J., Lijnen, H. R., and
Arnout, J., eds.) Leuven University Press, Leuven, pp. 227-265.
20.Takada, A., and Takada, Y. (1988)
Haemostasis, 18 (Suppl. 1), 25-35.
21.Bangert, K., Johnsen, A., Christensen, U., and
Thorsen, S. (1993) Biochem. J., 291, 623-625.
22.Kimura, S., and Aoki, N. (1986) J. Biol.
Chem., 261, 15591-15595.
23.Weisel, J. W., Veklich, Yu., Collet, J.-P., and
Francis, C. W. (1999) Thromb. Haemost., 82, 277-282.
24.Sakharov, D. V., Plow, E. F., and Rijken, D. C.
(1997) J. Biol. Chem., 272, 14477-14482.
25.Sottrup-Jensen, L., Stepanic, T. M., Kristensen,
T., Wierzbicki, D. M., Jones, C. M., Lonblad, P. B., Magnusson, S., and
Petersen, T. E. (1984) J. Biol. Chem., 259,
8318-8327.
26.Bajzar, L., Nesheim, M. E., and Tracy, P. B.
(1996) Blood, 88, 2093-2100.
27.Broze, G. J., Jr., and Higuichi, D. A. (1996)
Blood, 88, 3815-3823.
28.Wang, W., Boffa, M. B., Bajzar, L., Walker, J.
B., and Nesheim, M. E. (1998) J. Biol. Chem., 273,
27176-27181.
29.Eaton, D. L., Malloy, B. E., Tsai, S. P., Henzel,
W., and Drayana, D. (1991) J. Biol. Chem., 269,
21833-21838.
30.Bajzar, L., Morser, J., and Nesheim, M. E. (1996)
J. Biol. Chem., 271, 16603-16608.
31.Mao, S. S., Cooper, C. M., Wood, T., Shafer, J.
A., and Gardell, S. J. (1999) J. Biol. Chem., 274,
35046-35052.
32.Boffa, M. B., Wang, W., Bajzar, L., and Nesheim,
M. E. (1998) J. Biol. Chem., 273, 2127-2135.
33.Dahlbäck, B. (1995) Thromb. Res.,
77, 1-43.
34.Mosnier, L. O., Meijers, J. C. M., and Bouma, B.
N. (2001) Thromb. Haemost., 85, 5-11.
35.Strömqvist, M., Schatteman, K., Leurs, J.,
Verkerk, R., Andersson, J. O., Johansson, T., Scarpé, S., and
Hendrika, D. (2001) Thromb. Haemost., 85, 12-17.
36.Mosnier, L. O., von Dem Borne, P. A., Meijers, J.
C., and Bouma, B. N. (1998) Thromb. Haemost., 80,
829-835.
37.Chetaille, P., Alessi, M. C., Kouassi, D.,
Morange, P. E., and Juhan-Vague, I. (2000) Thromb. Haemost.,
83, 902-905.
38.Nesheim, M. E., Wang, W., Boffa, M. B.,
Nagashima, M., Morser, J., and Bajzar, L. (1997) Thromb.
Haemost., 78, 386-391.
39.Klement, P., Liao, P., and Bajzar, L. (1999)
Blood, 94, 2735-2743.
40.Nagashima, M., Werner, M., Wang, W., Zhao, L.,
Light, D. R., Pagila, R., Morser, J., and Verhallen, P. (2000)
Thromb. Res., 98, 333-342.
41.Refino, C. J., DeGuzman, L., Schmitt, D., Smyth,
R., Jeet, S., Lipari, M. T., Eaton, D., and Bunting, S. (2000)
Fibrinolysis Proteolysis, 14, 305-313.
42.Colucci, M., D'Aprile, A. M., Italia, A.,
Gresele, P., Morser, J., and Semeraro, N. (2001) Thromb.
Haemost., 85, 661-666.
43.Bajzar, L. (2000) Arterioscler. Thromb. Vasc.
Biol., 20, 2511-2518.
44.Van Tilburg, N. H., Rosendaal, F. R., and
Bertina, R. M. (2000) Blood, 95, 2855-2859.
45.Van Thiel, D. H., George, M., and Fareed, J.
(2001) Thromb. Haemost., 85, 667-670.
46.Meijers, J. C. M., Oudijk, E. J. D., Mosnier, L.
O., Bos, R., and Bouma, B. N. (2000) Br. J. Haematol.,
108, 518-523.
47.Badimon, J. J., Zaman, A., Helft, G., Fayad, Z.,
and Fuster, V. (1999) Thromb. Haemost., 82, 997-1004.
48.Butenas, S., and Mann, K. G. (2002)
Biochemistry (Moscow), 67, 3-12.
49.Meade, T. W., Ruddock, V., Stirling, Y.,
Charkrabarti, R., and Miller, G. J. (1993) Lancet, 342,
1076-1079.
50.Ridker, P. M. (1997) Thromb. Haemost.,
78, 53-59.
51.Thörgersen, A. M., Jansson, J. H., Boman,
K., Nilsson, T. K., Weinehall, L., Huhtasaari, F., and Hallmans, G.
(1998) Circulation, 98, 2241-2247.
52.Lowe, G. D. O., Yarmell, J. W. G., Sweetman, P.
M., Rumley, A., Thomas, H. F., and Elwood, P. C. (1998) Thromb.
Haemost., 79, 129-133.
53.Van der Bom, J. G., de Knijff, P., Haverkate, F.,
Bots, M. L., Meijer, P., de Jong, P. T. V. M., Hofman, A., Kluft, K.,
and Grobbee, D. E. (1997) Circulation, 95, 2623-2627.
54.Juhan-Vague, I., and Alessi, M. C. (1997)
Thromb. Haemost., 78, 656-660.
55.Sorokin, E. V., Karpov, Ju. A., Dobrovolsky, A.
B., Vilchinskaja, M. Ju., Panchenko, E. P., Titaeva, E. V., and
Kagan-Ponomarev, M. Ja. (1996) Ter. Arkhiv, 68,
19-23.
56.Kagan-Ponomarev, M. Ja., Dobrovolsky, A. B.,
Staroverov, I. I., Titaeva, E. V., Kravets, A. M., Pomerantsev, E. V.,
and Ruda, M. Ja. (1994) Kardiologiya, 34, 4-10.
57.Paganelli, F., Alessi, M. C., Morange, P.,
Maixent, J. M., Lévy, S., and Juhan-Vague, I. (1999) Thromb.
Haemost., 82, 104-108.
58.Kurnik, P. B. (1995) Circulation,
91, 1341-1346.
59.Panchenko, E., Dobrovolsky, A., Davletov, K.,
Titaeva, E., Podinovskaya, J., and Karpov, Yu. (1995) Eur. Heart
J., 16, 38-42.
60.Panchenko, E. P., Dobrovolsky, A. B., Davletov,
K. K., Titaeva, E. V., Skvortsov, A. V., Khrustaleva, O. P.,
Podinovskaya, J. A., and Karpov, Yu. A. (1995) Kardiologiya,
35, 18-23.
61.Van der Bom, J. G., Bots, M. L., Haverkate, F.,
Meyer, P., Hofman, A., Grobbee, D. E., and Kluft, C. (1999) Thromb.
Haemost., 81, 275-280.
62.Komarov, A., Panchenko, E., Dobrovolsky, A.,
Deev, A., Davletov, K., Eshkeeva, A., Markova, L., Titaeva, E., and
Karpov, Yu. (2001) Thromb. Haemost., Suppl., July, Abst.
P2808.
63.Salomaa, V., Stinson, V., Kark, J. D., Folsom, A.
R., Davis, C. E., and Wu, K. K. (1995) Circulation, 91,
284-290.
64.Thompson, S. G., Kienast, J., Pyke, S. D. M.,
Haverkate, F., and van de Loo, J. C. W. (1995) N. Engl. J. Med.,
332, 635-641.
65.Chandler, W. L., Alessi, M. C., Aillaud, M. F.,
Henderson, P., Vague, P., and Juhan-Vague, I. (1997)
Circulation, 96, 761-768.
66.Wiman, B., Andersson, T., Hallqvist, J.,
Reuterwall, C., Ahlbom, A., and deFaire, U. (2000) Arterioscler.
Thromb. Vasc. Biol., 20, 2019-2023.
67.Antiplatelet Trialist's Collaboration (1994)
Br. Med. J., 308, 81-106.
68.A Randomised, Blinded, Trial of Clopidogrel
Versus Aspirin in Patients at Risk of Ischemic Events (CAPRIE) (1996)
Lancet, 348, 1329-39.
69.Coller, B. S. (2001) Thromb. Haemost.,
86, 427-441.
70.Dugina, T. N, Kiseleva, E. V., Chistov, I. V.,
Umarova, B. A., and Strukova, S. M. (2002) Biochemistry
(Moscow), 67, 65-74.
71.Fareed, J., Hoppensteadt, D. A., and Bick, R. L.
(1998) Lectures XV Int. Congr. Thrombosis (Ulutin, O. N., ed.)
Antalya, pp. 163-181.
72.Gratsiansky, N. A. (1997) Klinich. Farmakol.
Terapiya, 6, 1-4.
73.Fenton, J. W., II, Jeske, W. P., Catalfamo, J.
L., Brezniak, D. V., Moon, D. G., and Shen, G. X. (2002)
Biochemistry (Moscow), 67, 85-91.
74.Aikawa, M., Rabkin, E., Okada, Y., Voglic, S. J.,
Clinton, S. K., Brinkerhoff, C. E., Sukhova, G. K., and Libbi, P.
(1998) Circulation, 97, 2433-2444.
75.Solovjeva, N. I. (1998) Bioorg. Khim.,
24, 245-255.
76.Lijnen, H. R. (2002) Biochemistry
(Moscow), 67, 92-98.