Gene Targeting of Tissue Factor, Factor X, and Factor VII in Mice: Their Involvement in Embryonic Development
M. Aasrum and H. Prydz*
Biotechnology Center of Oslo, P.O.B. 1125 Blindern, N-0317 Oslo, Norway; fax: 4722840501; E-mail: hans.prydz@biotek.uio.no* To whom correspondence should be addressed.
Received March 30, 2001; Revision received April 23, 2001
Inactivation of specific genes in mammals by gene targeting has accelerated our ability to determine gene function. Nearly all genes involved in the blood coagulation system have been knocked out in mice. Tissue factor (TF) is the main initiator of the coagulation system and functions as a cell surface receptor for coagulation factor VII (FVII). Knockout studies have shown that TF deficiency results in lethality around embryonic day (E) 8.5-10.5. The results suggest a role for TF in embryonic blood vessel development and maintenance of vascular integrity in the yolk sac. In addition, TF may be involved in the maintenance of the placental labyrinth. Factor X (FX) deficiency causes partial embryonic lethality between E11.5-12.5. FX-/- mice that were born died from fatal neonatal bleeding. In contrast, FVII deficiency is not embryonic lethal, but FVII-/- neonates died from hemorrhage within the first days after birth. The various lethal phenotypes of deficiencies of the different coagulation factors suggest involvement in processes beyond hemostasis. Both TF/FVIIa and FXa can trigger intracellular signaling events in certain cell types. Signaling by coagulation proteases and protease-activated receptors (PARs) may have important roles in embryonic development.
KEY WORDS: coagulation, gene targeting, tissue factor, factor VII, factor X, embryonic development, vasculogenesis
Abbreviations: TF) tissue factor; FVII) factor VII; FX) factor X; FV) factor V; FVIII) factor VIII; FIX) factor IX; FXI) factor XI; PAR) protease-activated receptor; MAPK) mitogen-activated protein kinase; BTEB2) basic transcription element binding protein 2; ETR101) eukaryotic transcription factor 101; TTP) tristetraproline; egr-1) early growth response gene-1; CTGF) connective tissue growth factor; AR) amphiregulin; EGF) epidermal growth factor; FGF) fibroblast growth factor; IL) interleukin; LIF) leukemia inhibitory factor; MIP) macrophage inflammatory protein; uPAR) urokinase plasminogen activator receptor; PAI) plasminogen activator inhibitor; PGE2R) prostaglandin E2 receptor.
Hemostasis initiated by injury to the vascular wall constitutes a major
defense system of our body. Following vessel wall injury, the
initiation, amplification, and control of hemostasis depend on
structurally unrelated membrane associated receptors and their ligands.
The coagulation system can be viewed as a cascade of proteolytic
reactions where zymogens are cleaved to produce active proteins [1]. The major players are the serine proteases of the
blood coagulation system such as factor VIIa (FVIIa), factor IXa
(FIXa), factor Xa (FXa), factor XIa (FXIa), and thrombin. These
proteins serve to induce and balance the hemostatic process by binding
to specific cell surface receptors on platelets, leukocytes, and
endothelium. The receptors in this system include tissue factor (TF),
thrombin receptor (PAR-1), and thrombomodulin. Following vascular
injury, TF is exposed to the circulation and the extracellular part of
TF serves as a receptor for FVII. This allows activation of the zymogen
FVII to an active serine protease (FVIIa). The TF-FVIIa complex
triggers the formation of both FXa and FIXa. With its cofactor, Factor
Va (FVa), FXa will then convert prothrombin to thrombin. Thrombin
promotes thrombus formation by activating platelets and converting
fibrinogen to fibrin, the major structural component of a blood clot.
Once trace amounts of thrombin are generated, there is a marked
amplification through thrombin feedback activation of factors V, VIII,
and XI.
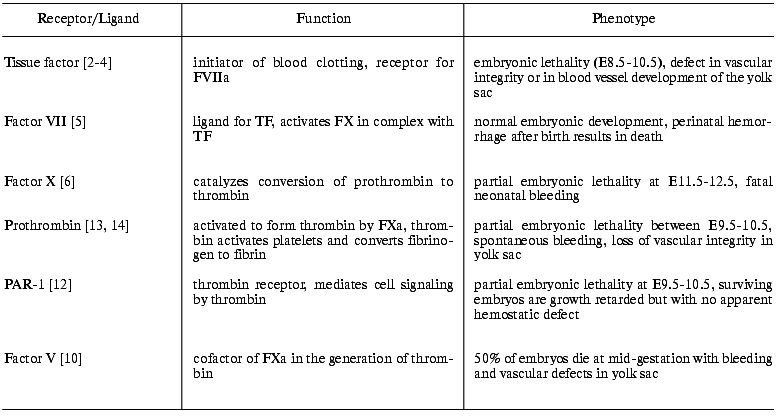
Nearly all of the major hemostasis-related genes have been knocked out in mice, including TF [2-4], FVII [5], FX [6], FIX [7], FVIII [8], von Willebrand factor [9], FV [10], FXI [11], PAR-1 [12], prothrombin [13, 14], and fibrinogen [15]. In addition, many of the genes encoding the known suppressors and inhibitors of the coagulation system have been disrupted. These studies have demonstrated the critical involvement of the coagulation factors in vascular development and maintenance of the vascular integrity during embryonic development (table). In this review, we shall summarize the effects of knocking out TF, FVII, and FX. In addition, some of the advances that have been made in the field of genetic manipulation of mice will be reviewed.
Functions of some protease receptors and ligands of the coagulation
system and the consequences of complete deficiency in mice
KNOCKING OUT GENES
The recently developed ability to inactivate specific genes in the mouse has accelerated the understanding of molecular and cellular aspects of normal and disease processes in a living animal. By the available techniques, it is possible to develop murine models for human diseases or determining the role of single genes. The knockout technique first involves homologous recombination in mouse embryonic stem cells (ES cells) by using a DNA construct containing an inactive copy of the gene to be knocked out. Second, these ES cells are incorporated into the developing mouse embryo to form germ-line chimera. By breeding these chimeric mice, the mutation made in ES cells can be transmitted to the next generation. Animals produced by this method will have the gene knocked out from the first step in development.
Many genes have been knocked out by the conventional knockout technique, and their functions have been discovered. However, the results have also shown the extreme complexity of genetic determination in mammals. Knockout studies do not always give clear answers due either to lack of phenotype or to the presence of ambiguous phenotypes. Many genes have multiple functions, and simple inactivation of such genes has limitations in demonstrating the complete spectrum of functions. In several studies, e.g., of the factors involved in the coagulation system, an early lethal phenotype has blocked studies of gene function at later stages in development or in the adult animal (table). Knocking out these genes results in complete or partial embryonic lethality, with the rest of the neonates dying a short while after birth.
By using the newly developed conditional knockout techniques, these problems can be overcome. In conditional knockouts, a specific gene can be targeted in a specific cell-type and/or at a specific stage in development [16]. This technique involves use of the Cre-loxP recombination system. Cre is a recombinase isolated from bacteriophage P1 that catalyzes a recombination between two 34-bp sequences referred to as loxP [17]. When Cre is expressed, any DNA between two loxP sites will be excised. By using a mouse strain that expresses Cre in a specific cell-type, it is possible to delete a gene in only this tissue. Two mouse strains are needed, the first one expressing Cre, the second one having loxP sites flanking the gene to be deleted. By crossing these lines, the gene will be deleted only in the tissue expressing Cre. The choice of a good tissue-specific promoter in this strategy is important. The promoter must be strong enough to give high expression of Cre. Many different tissue-specific promoters have been characterized by cell culture studies and in transgenic animals. Several promoters have been coupled to the Cre gene to control its expression pattern in mice. Temporal inactivation of genes is also possible by using inducible promoters to control the expression. Several inducible systems have been worked out including tamoxifen-, glucocorticoid-, tetracycline-, and heat shock induction of Cre [18-21]. A number of strains are available through the “Cre transgenic database” [22]. This database contains different Cre transgenic lines; both published and unpublished lines are included. Interesting promoters like the TIE-1 [23], TIE-2 [24], and SM22 [25] promoters have been used to control Cre expression. TIE-1 and TIE-2 are endothelial cell-specific genes and are important in the formation of blood vessels [26]. The SM22 promoter is specific for smooth muscle cells. In addition, to be restricted to smooth muscle cells, Cre is also regulated by tamoxifen induction in this mouse strain. In further studies of genes involved in development of the vascular system, these lines may be helpful.
TISSUE FACTOR
Biology of tissue factor. Tissue factor (TF) is believed to be the physiological initiator of blood coagulation. Mature TF is a 263 amino acid transmembrane glycoprotein consisting of a 219 amino acid extracellular domain, a 23 amino acid transmembrane segment, and a 21 amino acid cytoplasmic tail [27]. Cloning of the gene revealed structural similarities to the class II cytokine receptor superfamily. TF is a true receptor. Binding of its ligand FVIIa triggers intracellular calcium mobilization through phosphatidylinositol-specific phospholipase C [28, 29], as well as activation of mitogen-activated protein kinases (MAPKs) [30]. Recent studies have shown that binding of FVIIa to TF induces gene expression [31, 32]. The responding genes include transcriptional regulators, growth factors, proinflammatory cytokines, genes involved cellular reorganization/migration, and others.
TF is constitutively expressed by a variety of cells, most prominently in epithelial and perivascular cells. In this way TF creates a “hemostatic envelope” protecting against blood loss in trauma [33]. TF is normally absent from vascular endothelium and peripheral blood cells, but expression in monocytes and endothelium can be induced by cytokines and inflammatory molecules [34].
Human and murine genomic TF have been cloned and mapped to human chromosome 1 and murine chromosome 3, respectively [35, 36]. The structure of the two genes was found to be very similar, both organized into six exons spanning 12-13 kb. There are no reports of TF deficiency in humans. Regulation of the human TF gene has been characterized extensively in cell culture studies [37]. Sp1 mediates basal expression whereas c-Fos/c-Jun, c-Rel/p65, and Egr-1 mediate inducible expression. Enhancer elements responsible for the constitutive and inducible expression lie within the 300-400 bp of the promoter. Less is known about the regulatory regions that confer cell-type specific and inducible TF expression in vivo.
Recent studies have suggested that TF plays a nonhemostatic role in development and/or maintenance of blood vessels, resulting in death for deficient embryos [2-4]. TF has also been proposed to play a role in tumor-associated angiogenesis, metastasis, and inflammation [38, 39].
Involvement in embryonic blood vessel development. During embryonic development, blood vessels are initially formed by differentiation of mesodermal precursors (angioblasts) into vascular endothelial cells, followed by their assembly into a primary vascular network, a mechanism known as vasculogenesis [40]. This primary vascular plexus is then remodeled into the large and small vessels of the mature system by the process of angiogenesis [41]. Endothelial cells can initiate, but not complete the process of angiogenesis; periendothelial cells are essential for vascular maturation.
Pericytes/smooth muscle cells accumulate and differentiate around the endothelial cells. They inhibit proliferation and migration, and induce differentiation, quiescence, and survival of endothelial cells. In the developing embryo, blood cell formation and vasculogenesis begins at E7.5 in the blood islands of the yolk sac [42]. The endothelial cells and hematopoetic cells of these islands are proposed to originate from a common precursor, termed the hemangioblast. The primary vascular plexus formed in the yolk sac will form the large vitelline blood vessel.
Three independent groups have reported that TF-/- embryos die between E8.5-10.5, because of the inability to establish or maintain vascular integrity and defective yolk sac vessel development [2-4]. Mice heterozygous for the inactivated TF allele were phenotypically normal. No morphological defects were observed in TF-/- embryos at E8.5. At E9.5 wasting of TF-/- embryos was observed with variable onset. Approximately 90% of the TF-/- embryos were dead at embryonic day 10.5. At this stage they were growth retarded and pale, with abnormalities in the yolk sac. In TF deficient yolk sacs, the mesenchyme-derived smooth muscle cells/pericytes fail to accumulate and differentiate around endothelial cell-lined capillaries [2]. The blood vessels become fragile, and blood is leaking into the extravascular cavity.
The blood circulation from yolk sac to the growing embryo is nearly inhibited. The capillary plexus of the yolk sac is enlarged and disordered, and the vitelline vessels are not properly developed. At the time when the TF-/- embryos die, the yolk sac vasculature is completed in TF+/+ embryos. The extra-embryonic and embryonic vasculatures are fused, and the embryonic blood circulation begins. The nutrition of the embryo depends on the yolk sac vasculature. Wasting of the TF-/- embryo appears to be a result of the lack of essential nutrients due to a defective yolk sac vasculature.
In a 129/SvJ or a 129/SvJ:NIH Black Swiss background no TF-/- embryos have survived longer than E10.5. If must be mentioned that a few TF-/- embryos have survived beyond E10.5 in a 129/SvJ:C57BL/6 genetic background [43]. The small number of embryos survived into late gestation, and died then due to hemorrhage. The genetic background has an influence on this phenotype, but what is causing this compensatory effect is not known.
To further investigate the role of TF during embryogenesis, transgenic mice expressing a human TF minigene lacking the intracellular domain (tailless) or with a mutation in the extracellular domain were made [44]. These transgenic mice were then crossed with mice heterozygous for the mouse TF gene (TF+/-) to create offspring heterozygous for both the transgene construct and the mouse TF gene. Rescue experiments were then performed by crossing these offspring with TF+/- mice. Pups were sacrificed and genotyped.
Transgenic mice expressing the tailless human TF mutation rescued the embryonic lethality of murine TF-/- embryos. In contrast, the human TF extracellular domain mutant did not rescue the embryos. These results indicated that the intracellular domain is not required for embryonic development. However, binding of FVIIa to the extracellular domain of TF and formation of a proteolytically active TF/FVIIa complex is required for mouse embryogenesis.
Involvement in uterine hemostasis and maintenance of the placental labyrinth. TF deficient mice are rescued when expressing low levels of a human TF minigene [45]. The expression of the minigene was only about 1% relative to mouse TF, but embryos developed normally with no signs of bleeding. The low TF level can maintain hemostasis and normal development. Adult low-TF mice are fertile. When low-TF female mice are bred with low-TF or wild type males, fatal postpartum uterine hemorrhage is reported, independent of the genotype of the pup. Fatal mid-gestational hemorrhage was also observed, a result of low levels of embryonically derived TF in the placenta [46]. Maternal blood pools and placental hemorrhage were observed in the labyrinth layer between E12 and E16 in low-TF embryos. Further examination of these embryos showed a thinning of the layer I trophoblasts that normally forms a continuous lining of the maternal blood space during gestation. This suggests a possible role for TF in maintenance of the placenta, but the mechanism involved in this process is not clear.
In deficiencies of other coagulation factors similar phenotypes are reported with defects in blood vessel development. Approximately 50% of FV deficient embryos die at mid-gestation with bleeding and vascular abnormalities in the yolk sac [10]. Embryos that are born suffer from fatal postnatal hemorrhage. It has been reported that prothrombin deficiency also leads to defective yolk sac vasculature at mid-gestation [13]. More than 50% of the embryos die before E10.5.
The lethality of TF deficient embryos is more severe compared to the other coagulation factors, suggesting additional functions of TF beyond its involvement in the coagulation process. It remains to be determined whether TF acts in a FVII-dependent or -independent manner. Currently FVII is the only known ligand for TF, but TF may interact with other ligands or receptors and take part in different processes during embryonic development. The alternative hypothesis is that TF may be involved in embryonic development by thrombin generation. Approximately 50% of embryos deficient in thrombin receptor, PAR-1 (protease-activated receptor 1), die at E10.5 [12]. There is a possible link between TF-dependent activation of coagulation and the PAR-1 signaling system. Generation of thrombin may lead to activation of the PAR-1-dependent intracellular signaling during embryogenesis, more specifically in the visceral yolk sac.
FACTOR VII
The factor VII protein. FVII is a vitamin K-dependent serine protease zymogen synthesized in the liver. The gene is organized into 8 exons and is located on chromosome 13 in humans and chromosome 8 in mice. Both genes are cloned and characterized [47, 48]. The gene is located centromeric of FX in both humans and mice. The human FVII is positively regulated primarily by a liver-specific transcription factor, hepatocyte nuclear factor-4 (HNF-4) [49]. The mature protein consists of 406 residues, but is synthesized with a 38-residue leader sequence directing the polypeptide to rough endoplasmic reticulum. FVII is a zymogen and requires activation to get biological activity. FVII is activated to FVIIa by cleavage of its Arg152-Ile153 bond. After cleavage, a single disulfide bond connects the two resulting chains. This activation process requires Ca2+, phospholipids, and interaction with TF. The TF/FVIIa complex has two substrates in the coagulation process, FIX and FX. FVII deficiency caused either by reduced production or activity of the protein normally leads to increased bleeding tendency [50].
Factor VII deficient mice. The FVII gene was inactivated by deleting the entire coding sequence for the mature protein [5]. The FVII-/- embryos developed normally, but were under-represented according to the expected Mendelian frequency at birth (observed 18.4%, expected 25%). Timed matings between FVII+/- males and females were used to analyze the frequency of FVII-/- embryos at various stages during gestation (E9.5-E18.5). FVII deficient embryos were present at their expected frequency (25%) at each of the stages. The under-representation at birth was suggested to be a result from death and consumption from their parents. No vascular defects were seen in yolk sac or in the embryo itself, in contrast to the defects in TF deficient embryos. Measurement of FVII plasma proteolytic activity from deficient embryos at various stages demonstrated that there was no detectable FVII activity. The proteolytic activity of FVII in heterozygous embryos was approximately 50% compared to age-matched wild type embryos In wild type embryos FVII expression was minimal at E9.5 in both the embryo and yolk sac but abundant in neonates. More than half of the FVII-/- embryos died because of fatal intra-abdominal bleeding within the first 24 h after birth, whereas most of the remaining mice died from intracranial hemorrhage before the age of 24 days. Analysis of the embryos showed widespread extravasation of red blood cells into the peritoneal cavity. The blood vessel development was not affected. There is a possibility that the normal development of FVII deficient embryos is dependent on transfer of maternal FVII. Humans with FVII levels down to approximately 1% survive. Therefore, low levels of maternally transferred FVII could lead to normal embryonic development. If any such maternal transfer exists, the level is below the present detection limit.
FACTOR X
The factor X protein. FX like FVII is a vitamin K-dependent serine protease zymogen synthesized in the liver [51]. The 488-amino acid FX precursor is modified to a mature two-chain zymogen by gamma-carboxylation and cleavage of the pre-pro-sequence. The FX gene is located downstream (telomeric) of the FVII gene. Like the other vitamin K-dependent coagulation factors, the FX gene is divided into 8 exons coding for 8 different functional domains. The promoter region of the murine FX gene contains binding sites for the transcription factors HNF-4, NF-Y (nuclear factor Y), and GATA-4. These sites have influence on the transcriptional activity of the gene [52]. FX is converted to its active form FXa by the TF/FVIIa complex during the initiation phase of blood coagulation. In association with FVa, FXa forms the prothrombinase complex catalyzing the conversion of prothrombin to thrombin. Hereditary FX deficiency is a rare autosomal recessive disorder, but the genetic defects underlying the deficiencies are largely uncharacterized.
FX participates in other processes beyond coagulation. Like FVIIa, FXa induces intracellular calcium signals in certain cells [28, 29]. The signaling is independent of thrombin generation. The protein acts as a mitogenic and signaling agent in vascular smooth muscle cells [53], and it stimulates an acute inflammatory response [54]. In addition, FX has been shown to induce cytokine production and expression of adhesion molecules by human umbilical vein endothelial cells [55].
Factor X deficient mice. The FX gene was inactivated by deleting an 18 kb genomic fragment containing the entire coding part of the gene [6]. Homozygous FX-deficient embryos were underrepresented among offspring of heterozygous breeding pairs. Only 15% of the neonates were FX-/-, and 50% of them were dying due to fatal bleeding the first day after birth. The majority of the remaining embryos were dying within 5 days. Microscopic analysis confirmed widespread bleeding in the peritoneal cavity, but it did not reveal any abnormalities in organ or blood vessel development. Timed matings showed an expected ratio of FX-/- embryos at E9.5. At E12.5 the ratio of FX-/- was reduced from 25 to 17%. At these stages the surviving embryos were grossly indistinguishable from their wild-type littermates. However, some embryos showed signs of bleeding. The portion of FX-/- remained constant at 16% after E12.5. Thus, the FX deficiency leads to partial embryonic lethality between E11.5 and E12.5. The FX deficiency resembles the phenotype of FV and prothrombin deficiency, suggesting that generation of thrombin provides a critical embryonic function at mid-gestation. In contrast to FX deficiency, yolk sac abnormalities are reported in FV and prothrombin deficiency [10, 14]. However, potential vascular defects occurring between E11.5-12.5 cannot be excluded, as degeneration and necrosis prevented further studies.
It seems unlikely that the essential function of FX in the embryos involves clot formation since fibrinogen deficient embryos develop normally [15]. A possible role for FX during development can be the FV/FX activation of prothrombin, leading to thrombin-mediated intracellular signaling by the thrombin receptor PAR-1. In PAR-1 deficiency, 50% of the embryos die between embryonic day 9.5 and 10.5. PAR-1 is expressed by vascular endothelial cells and smooth muscle cells [56, 57] and is thus well positioned to mediate communication between the blood and cells of the vessel wall. Signaling in endothelial cells is important for normal vascular development. No vascular abnormalities have been reported in PAR-1-/- mice and they have normal skin wound healing [58]. However, it is reported that PAR-1-/- mice have altered responses to arterial injury [59].
SIGNALING BY THE TF/FVIIa COMPLEX AND FXa
When FVIIa binds to TF on various cell types, intracellular Ca2+ oscillations are induced through phosphatidylinositol-specific phospholipase C [28, 29]. The complex induces signaling via different MAPKs including p38, extracellular signal-regulated kinase 1/2 (Erk1/2), and c-Jun N-terminal kinase, and upregulation of the early growth response gene (egr-1) [30].
In recent studies using the cDNA macroarray technology we have shown that the signaling through the MAPK pathways p38 and Erk1/2 induces gene expression in keratinocytes [31]. The pattern of genes induced may suggest a role for TF/FVIIa signaling in wound repair. Several of these genes are involved in the earliest steps of the wound reaction and may also mediate other effects of TF in metastasis and tumor angiogenesis. Genes that were up-regulated included transcription factors (c-fos, ETR101, fra-1, c-myc, BTEB2, TTP, and egr-1), growth factors (CTGF, AR, hbEGF, and FGF-5), proinflammatory cytokines (IL-8, IL-1beta, LIF, and MIP2alpha), and others (RhoE, uPAR, collagenase 1 and 3, PAI-2, Jagged1, cyclophilin, GADD45, and PGE2R). Another study showed that FVIIa induced expression of Cyr61 and CTGF in human fibroblasts [32]. Upregulation of VEGF (vascular endothelial growth factor) in fibroblasts has also been reported [60]. VEGF production is induced in fibroblasts when FVIIa binds to TF, but the process involves generation of FXa and thrombin [61]. Taken together these data suggest that the formation of TF/FVIIa complex may affect various biological processes by inducing expression of downstream effector proteins.
Both FVIIa and FXa must be proteolytically active to generate Ca2+ signals. This suggested that protease-activated receptors were involved in the signaling process. PAR-1 is an important mediator of thrombin signaling [62]. In addition, three more PARs have been identified. Like PAR-1, PAR-3 and PAR-4 can be activated by thrombin [63, 64]. In contrast, PAR-2 is not activated by thrombin. The PARs belong to a subclass of G-protein-coupled receptors, and they are activated by proteolysis of a specific peptide bond in the extracellular region of the protein. They are expressed widely in intra- and extravascular cells, and signaling by these receptors in response to injury is expected to regulate diverse cellular functions including gene expression, cell proliferation, and migration.
Like thrombin, FVIIa and FXa can trigger signaling events in certain cells. Studies in Madin-Darby canine kidney cells showed that PAR-1 was not involved in the generation of the Ca2+ signal by FVIIa induction [29]. TF/FVIIa-induced signal transduction leads to Erk1/2 MAPK phosphorylation and requires proteolytic activity of FVIIa [30, 65, 66]. Exposure of TF-transfected baby hamster kidney (BHK) cells to FXa did not induce Erk1/2 phosphorylation. In addition, a specific inhibitor of FXa, recombinant tic anticoagulant protein, did not inhibit FVIIa-induced MAPK activation; neither did inhibition of thrombin. This excludes an indirect route for signal transduction via known or unknown thrombin and FXa receptors.
The cytoplasmic domain of TF is not required for the Erk1/2 MAPK activation. Recent studies of TF-transfected BHK cells suggest that FVIIa-induced intracellular signaling does not involve the activation of a known PAR, and that the intracellular activity induced by FVIIa is distinctly different from that induced by other serine proteases [67].
Recent studies have suggested that TF-dependent activation of FXa can trigger signaling events independent of thrombin generation, and that PAR-2 may function as a FVIIa/FXa receptor [68]. PAR-2 may be activated directly by TF/FVIIa and/or indirectly by TF/FVIIa-generated FXa. FVIIa can activate PAR-2 in the presence of TF and FX in keratinocytes and endothelial cells. When FVIIa binds to TF, this complex converts inactive zymogen FX to the active protease FXa. FXa can then cleave PAR-2 to trigger transmembrane signaling. The signaling through PAR-2 may be important in inflammation and in establishing a link between coagulation and inflammation. Several studies implicate PAR-2 in tissue injury and inflammation. PAR-2 expression is upregulated by inflammatory mediators such as TNF-alpha and IL-1 [69]. PAR-2 is expressed in vascular endothelial cells, where it mediates proliferation, and in vascular smooth muscle cells.
PAR-2 deficient mice have been made [70]. At birth, PAR-2-/- embryos were underrepresented from the expected Mendelian ratio (16.7% compared to 25%). Furthermore, 20% of the embryos were dead at birth, or found dead within 48 h. By anatomical and histological analysis of these pups, no abnormalities were observed. Surviving PAR-2-/- mice appeared normal, and homozygous matings gave normal litter sizes. Recent studies have shown that PAR-2 is an important receptor in inflammation and appears necessary for some of the earliest inflammatory responses in vivo [71].
PERSPECTIVE
The hemostatic proteases and their receptors provide important molecular links between blood coagulation and vascular cell functions. They appear to be indispensable during embryonic development for establishing the early vascular system.
Based on the similar phenotypes of mice deficient in FX, FV, and prothrombin, the failure of embryos during mid-gestation may be a result of a defect in a common pathway shared by these proteins. It is likely that the defect is due to a failure in thrombin signaling through PAR-1. Formation of fibrin, in itself, is not required for embryogenesis. The presence of FX and thrombin may be important for other functions than blood clotting related to the cell signaling processes in which they are involved.
Lethality is higher for TF deficient embryos compared to the level of lethality in FV-, FX-, and prothrombin deficiency. This may be due to additional functions of TF beyond its involvement in the coagulation cascade. TF deficiency may affect the regulation of protease generation and thereby inhibiting cell signaling processes required for embryonic development. Whether TF interacts with FVII or with a yet unidentified factor in these processes is unknown. Factor FVII deficient embryos develop normally but suffer from fatal hemorrhage after birth. These results suggest that TF may interact with another factor. Alternatively, placental transfer of FVII might rescue FVII deficient embryos.
Strategies to bypass the embryonic lethality of these deficiencies are possible by using tissue-specific or inducible knockout techniques by the Cre-loxP system. The deficiencies can then be studied later in development and in adult animals. Such mice are being prepared.
Work in the authors' laboratory has been supported by grants from The Norwegian Council on Cardiovascular Disease, The Research Council of Norway, and The Owren and The Jahre foundation to H. P.
REFERENCES
1.Davie, E. W. (1995) Thromb. Haemost.,
74, 1-6.
2.Carmeliet, P., Mackman, N., Moons, L., Luther, T.,
Gressens, P., van Vlaenderen, I., Demunck, H., Kasper, M., Breier, G.,
Evrard, P., Muller, M., Risau, W., Edgington, T., and Collen, D. (1996)
Nature, 383, 73-75.
3.Bugge, T. H., Xiao, Q., Kombrinck, K. W., Flick, M.
J., Holmback, K., Danton, M. J., Colbert, M. C., Witte, D. P.,
Fujikawa, K., Davie, E. W., and Degen, J. L. (1996) Proc. Natl.
Acad. Sci. USA, 93, 6258-6263.
4.Toomey, J. R., Kratzer, K. E., Lasky, N. M.,
Stanton, J. J., and Broze, G. J. (1996) Blood, 88,
1583-1587.
5.Rosen, E. D., Chan, J. C., Idusogie, E., Clotman,
F., Vlasuk, G., Luther, T., Jalbert, L. R., Albrecht, S., Zhong, L.,
Lissens, A., Schoonjans, L., Moons, L., Collen, D., Castellino, F. J.,
and Carmeliet, P. (1997) Nature, 390, 290-294.
6.Dewerchin, M., Liang, Z., Moons, L., Carmeliet P.,
Castellino, F. J., Collen, D., and Rosen, E. D. (2000) Thromb.
Haemost., 83, 185-190.
7.Kundu, R. K., Sangiorgi, F., Wu, L. Y., Kurachi,
K., Anderson, W. F., Maxson, R., and Gordon, E. M. (1998) Blood,
92, 168-174.
8.Bi, L., Lawler, A. M., Antonarakis, S. E., High, K.
A., Gearhart, J. D., and Kazazian, H. H. (1995) Nat. Genet.,
10, 119-121.
9.Denis, C., Methia, N., Frenette, P. S., Rayburn,
H., Ullman-Cullere, M., Hynes, R. O., and Wagner, D. D. (1998) Proc.
Natl. Acad. Sci. USA, 95, 9524-9529.
10.Cui, J., O'Shea, K. S., Purkayastha, A.,
Saunders, T. L., and Ginsburg, D. (1996) Nature, 384,
66-68.
11.Gailani, D., Lasky, N. M., and Broze, G. J.
(1997) Blood Coagul. Fibrinolysis, 8, 134-144.
12.Connolly, A. J., Ishihara, H., Kahn, M. L.,
Farese, R. V., and Coughlin, S. R. (1996) Nature, 381,
516-519.
13.Sun, W. Y., Witte, D. P., Degen, J. L., Colbert,
M. C., Burkart, M. C., Holmback, K., Xiao, Q., Bugge, T. H., and Degen,
S. J. (1998) Proc. Natl. Acad. Sci. USA, 95,
7597-7602.
14.Xue, J., Wu, Q., Westfield, L. A., Tuley, E. A.,
Lu, D., Zhang, Q., Shim, K., Zheng, X., and Sadler, J. E. (1998)
Proc. Natl. Acad. Sci. USA, 95, 7603-7607.
15.Suh, T. T., Holmback, K., Jensen, N. J.,
Daugherty, C. C., Small, K., Simon, D. I., Potter, S., and Degen, J. L.
(1995) Genes Dev., 9, 2020-2033.
16.Rajewsky, K., Gu, H., Kuhn, R., Betz, U. A.,
Muller, W., Roes, J., and Schwenk, F. (1996) J. Clin. Invest.,
98, 600-603.
17.Abremski, K., Wierzbicki, A., Frommer, B., and
Hoess, R. H. (1986) J. Biol. Chem., 261, 391-396.
18.Metzger, D., Clifford, J., Chiba, H., and
Chambon, P. (1995) Proc. Natl. Acad. Sci. USA, 92,
6991-6995.
19.Brocard, J., Feil, R., Chambon, P., and Metzger,
D. (1998) Nucleic Acids Res., 26, 4086-4090.
20.Utomo, A. R., Nikitin, A. Y., and Lee, W. H.
(1999) Nat. Biotechnol., 17, 1091-1096.
21.Dietrich, P., Dragastis, I., Xuan, S., Zeitlin,
S., and Efstratiadis, A. (2000) Mamm. Genome, 11,
196-205.
22.Nagy, A. (2001) Cre Transgenic Database.
www.mshri.ca/nagy/cre.htm.
23.Gustafsson, E., Brakebusch, C., Hietanen, K., and
Fassler, R. (2001) J. Cell Sci., 114, 671-676.
24.Kisanuki, Y. Y., Hammer, R. E., Miyazaki, J. J.,
Williams, S. C., Richardson, J. A., and Yanagisawa, M. (2001) Dev.
Biol., 230, 230-242.
25.Kuhbandner, S., Brummer, S., Metzger, D.,
Chambon, P., Hofmann, F., and Feil, R. (2000) Genesis,
28, 15-22.
26.Sato, T. N., Tozawa, Y., Deutsch, U.,
Wolburg-Buchholz, K., Fujiwara, Y., Gendron-Maguire, M., Gridley, T.,
Wolburg, H., Risau, W., and Qin, Y. (1995) Nature, 376,
70-74.
27.Morrissey, J. H., Fakhrai, H., and Edgington, T.
S. (1987) Cell, 50, 129-135.
28.Røttingen, J.-A., Enden, T., Camerer, E.,
Iversen, J.-G., and Prydz, H. (1995) J. Biol. Chem., 270,
4650-4660.
29.Camerer, E., Røttingen, J.-A., Iversen,
J.-G., and Prydz, H. (1996) J. Biol. Chem., 271,
29034-29042.
30.Camerer, E., Rottingen, J. A., Gjernes, E.,
Larsen, K., Skartlien, A. H., Iversen, J. G., and Prydz, H. (1999)
J. Biol. Chem., 274, 32225-32233.
31.Camerer, E., Gjernes, E., Wiiger, M., Pringle,
S., and Prydz, H. (2000) J. Biol. Chem., 275,
6580-6585.
32.Pendurthi, U. R., Allen, K. E., Ezban, M., and
Rao, L. V. (2000) J. Biol. Chem., 275, 14632-14641.
33.Drake, T. A., Ruf, W., Morrissey, J. H., and
Edgington, T. S. (1989) J. Cell Biol., 109, 389-395.
34.Camerer, E., Kolsto, A. B., and Prydz, H. (1996)
Thromb. Res., 81, 1-41.
35.Scarpati, E. M., Wen, D., Broze, G. J., Miletich,
J. P., Flandermeyer, R. R., Siegel, N. R., and Sadler, J. E. (1987)
Biochemistry, 26, 5234-5238.
36.Mackman, N., Imes, S., Maske, W. H., Taylor, B.,
Lusis, A. J., and Drake, T. A. (1992) Arterioscler. Thromb.,
12, 474-483.
37.Mackman, N. (1997) Thromb. Haemost.,
78, 747-754.
38.Zhang, Y., Deng, Y., Luther, T., Muller, M.,
Ziegler, R., Waldherr, R., Stern, D. M., and Nawroth, P. P. (1994)
J. Clin. Invest., 94, 1320-1327.
39.Bromberg, M. E., Konigsberg, W. H., Madisen, J.
F., Pawashe, A., and Garen, A. (1995) Proc. Natl. Acad. Sci.
USA, 92, 8205-8209.
40.Carmeliet, P. (2000) Nat. Med., 6,
389-395.
41.Risau, W. (1997) Nature, 386,
671-674.
42.Auerbach, R., Gilligan, B., Lu, L. S., and Wang,
S. J. (1997) J. Cell Physiol., 173, 202-205.
43.Toomey, J. R., Kratzer, K. E., Lasky, N. M., and
Broze, G. J. (1997) Proc. Natl. Acad. Sci. USA, 94,
6922-6926.
44.Parry, G. C., and Mackman, N. (2000) J. Clin.
Invest., 105, 1547-1554.
45.Parry, G. C., Erlich, J. H., Carmeliet, P.,
Luther, T., and Mackman, N. (1998) J. Clin. Invest., 101,
560-569.
46.Erlich, J., Parry, G. C., Fearns, C., Muller, M.,
Carmeliet, P., Luther, T., and Mackman, N. (1999) Proc. Natl. Acad.
Sci. USA, 96, 8138-8143.
47.Hagen, F. S., Gray, C. L., O'Hara, P., Grant, F.
J., Saari, G. C., Woodbury, R. G., Hart, C. E., Insley, M., Kisiel, W.,
and Kurachi, K. (1986) Proc. Natl. Acad. Sci. USA, 83,
2412-2416.
48.Idusogie, E., Rosen, E. D., Carmeliet, P.,
Collen, D., and Castellino, F. J. (1996) Thromb. Haemost.,
76, 957-964.
49.Pollak, E. S., Hung, H. L., Godin, W., Overton,
G. C., and High, K. A. (1996) J. Biol. Chem., 271,
1738-1747.
50.Cooper, D. N., Millar, D. S., Wacey, A., Banner,
D. W., and Tuddenham, E. G. (1997) Thromb. Haemost., 78,
151-160.
51.Hertzberg, M. (1994) Blood Rev., 8,
56-62.
52.Wilberding, J. A., and Castellino, F. J. (2000)
Thromb. Haemost., 84, 1031-1038.
53.Gasic, G. P., Arenas, C. P., Gasic, T. B., and
Gasic, G. J. (1992) Proc. Natl. Acad. Sci. USA, 89,
2317-2320.
54.Cirino, G., Cicala, C., Bucci, M., Sorrentino,
L., Ambrosini, G., DeDominicis, G., and Altieri, D. C. (1997) J.
Clin. Invest., 99, 2446-2451.
55.Senden, N. H., Jeunhomme, T. M., Heemskerk, J.
W., Wagenvoord, R., van't Veer, C., Hemker, H. C., and Buurman, W. A.
(1998) J. Immunol., 161, 4318-4324.
56.Muramatsu, I., Laniyonu, A., Moore, G. J., and
Hollenberg, M. D. (1992) Can. J. Physiol. Pharmacol., 70,
996-1003.
57.McNamara, C. A., Sarembock, I. J., Gimple, L. W.,
Fenton, J. W., Coughlin, S. R., and Owens, G. K. (1993) J. Clin.
Invest., 91, 94-98.
58.Connolly, A. J., Suh, D. Y., Hunt, T. K., and
Coughlin, S. R. (1997) Am. J. Pathol., 151,
1199-1204.
59.Cheung, W. M., D'Andrea, M. R., Andrade-Gordon,
P., and Damiano, B. P. (1999) Arterioscler. Thromb. Vasc. Biol.,
19, 3014-3024.
60.Ollivier, V., Bentolila, S., Chabbat, J., Hakim,
J., and de Prost, D. (1998) Blood, 91, 2698-2703.
61.Ollivier, V., Chabbat, J., Herbert, J. M., Hakim,
J., and de Prost, D. (2000) Arterioscler. Thromb. Vasc. Biol.,
20, 1374-1381.
62.Coughlin, S. R. (2000) Nature, 407,
258-264.
63.Ishihara, H., Connolly, A. J., Zeng, D., Kahn, M.
L., Zheng, Y. W., Timmons, C., Tram, T., and Coughlin, S. R. (1997)
Nature, 386, 502-506.
64.Xu, W. F., Andersen, H., Whitmore, T. E.,
Presnell, S. R., Yee, D. P., Ching, A., Gilbert, T., Davie, E. W., and
Foster, D. C. (1998) Proc. Natl. Acad. Sci. USA, 95,
6642-6646.
65.Poulsen, L. K., Jacobsen, N., Sorensen, B. B.,
Bergenhem, N. C., Kelly, J. D., Foster, D. C., Thastrup, O., Ezban, M.,
and Petersen, L. C. (1998) J. Biol. Chem., 273,
6228-6232.
66.Sorensen, B. B., Freskgard, P. O., Nielsen, L.
S., Rao, L. V., Ezban, M., and Petersen, L. C. (1999) J. Biol.
Chem., 274, 21349-21354.
67.Petersen, L. C., Thastrup, O., Hagel, G.,
Sorensen, B. B., Freskgard, P. O., Rao, L. V., and Ezban, M. (2000)
Thromb. Haemost., 83, 571-576.
68.Camerer, E., Huang, W., and Coughlin, S. R.
(2000) Proc. Natl. Acad. Sci. USA, 97, 5255-5260.
69.Nystedt, S., Ramakrishnan, V., and Sundelin, J.
(1996) J. Biol. Chem., 271, 14910-14915.
70.Damiano, B. P., Cheung, W. M., Santulli, R. J.,
Fung-Leung, W. P., Ngo, K., Ye, R. D., Darrow, A. L., Derian, C. K., de
Garavilla, L., and Andrade-Gordon, P. (1999) J. Pharmacol. Exp.
Ther., 288, 671-678.
71.Lindner, J. R., Kahn, M. L., Coughlin, S. R.,
Sambrano, G. R., Schauble, E., Bernstein, D., Foy, D., Hafezi-Moghadam,
A., and Ley, K. (2000) J. Immunol., 165, 6504-6510.