REVIEW: Contact System. New Concepts on Activation Mechanisms and Bioregulatory Functions
G. A. Yarovaya*, T. B. Blokhina, and E. A. Neshkova
Department of Biochemistry, Russian Medical Academy of Postgraduate Education, Barrikadnaya ul. 2/1, Moscow, 123995 Russia; fax: (095) 945-2415; E-mail: acadbio@orc.ru; yarov@aha.ru* To whom correspondence should be addressed.
Received July 3, 2001; Revision received September 18, 2001
This review considers the data of recent years concerning the contact system initiating the activation of blood plasma proteolytic systems, such as hemocoagulation, fibrinolysis, kininogenesis, and also complement and angiotensinogenesis. The main proteins of the contact system are the factors XII and XI, prekallikrein, and high-molecular-weight kininogen. The data on the structure, functions, and biosynthesis of these proteins and on their genes are presented. Studies in detail on the protein-protein interactions during formation of the ensemble of the contact system components on the anionic surface resulted in the postulation of the mechanism of activation of this system associated with generation of the XIIa factor and of kallikrein. This mechanism is traditionally considered a trigger of processes for the internal pathway of the hemocoagulating cascade. However, the absence of direct confirmation of such activation in vivo and the absence of hemorrhagia in the deficiency of these components stimulated the studies designed to find another mechanism of their activation and physiological role outside of the hemostasis system. As a result, a new concept on the contact system activation on the endothelial cell membrane was proposed. This concept is based on the isolation of a complex of proteins, which in addition to the above-mentioned proteins includes cytokeratin 1 and the receptors of the urokinase-like plasminogen activator and of the complement q-component. The ideas on the role of this system in the biology of vessels are developed. Some of our findings on the effect of leukocytic elastase on the key components of the contact system are also presented.
KEY WORDS: contact system, factor XII, factor XI, prekallikrein, kallikrein, high-molecular-weight kininogen, polyanionic surface, endothelial cells, ensemble of the contact system components, activity regulation
Blood coagulation starts immediately after a damage of the vascular endothelium and uncovering of the subendothelial structures. The trigger mechanism of the so-called “contact” phase of hemostasis activation is studied in detail on negatively charged surfaces, such as kaolin, sulfatides, dextran sulfates, phospholipids, urate crystals, etc. During the activation of proenzymes, the molecules are mutually oriented due to generation on the activating surface of an ensemble consisting of four proteins: hemocoagulation factors XII and XI, high-molecular-weight kininogen (HMK), and prekallikrein (PK) [1-4]. This results in the contact activation of factor XII and its conversion into factor XIIa [4, 5-7]. Factor XII can also be activated during proteolysis under the influence of blood plasma kallikrein, and this results in production of two active forms of factor XII--factor alphaXIIa and factor betaXIIa [1, 3, 8]. Concepts on the character and role of the contact activation of plasma proteolysis are now significantly changed and enlarged. The mechanism of factor XII contact activation that was postulated earlier suggested the involvement of this activation in the triggering of the reaction cascade of the internal pathway of hemocoagulation. The involvement of the kallikrein-kinin system (KKS) components in factor XII activation on a polyanionic surface was shown and the reciprocal mechanism of factor XII and PK activation was established [9, 10].
Later it became clear that activation of the blood plasma contact system plays an essential role in fibrinolysis because it results in the activation of plasmin and prourokinase [11, 12]. The proteinases generated after initiation of the contact system also influence the complement. Thus, plasmin, betaXIIa factor, and kallikrein are responsible for activation of the r- and s-subunits of the complement first component, which are precursors of serine proteinases of the classic activation pathway and factor B, which is a proform of the serine proteinase of the alternative pathway of complement activation. Moreover, kallikrein is an activator of prorenin [13]. Thus, the contact phase was shown to initiate the activation not only of the coagulation system but also of all other proteolytic systems of the blood plasma: KKS, complement, fibrinolytic, and the renin-angiotensin system [12-16].
During recent years, concepts on the mechanism of the activation of the contact system significantly changed because an alternative mechanism of contact system activation was found on endothelial cells.
This review considers data on the structure, functional properties, and mechanisms of interaction of proteins involved in the contact system and also the new concept on the activation of this system on the endothelial cell surface.
Moreover, some results are presented of our studies on the effect of leukocytic elastase on the key factors of the contact system in vitro using the purified proteins and in vivo in the blood plasma of patients with inflammatory diseases.
STRUCTURE AND FUNCTIONAL PROPERTIES OF THE CONTACT SYSTEM
FACTORS
High-molecular-weight kininogen. Human blood plasma contains two kininogens: high-molecular-weight and low-molecular-weight kininogens (HMK and LMK, respectively), and their synthesis is encoded by the same gene located in the third chromosome. According to Kitamura et al. [16], the kininogen gene contains eleven exons, nine of them forming three triplet exons each of which seems to originate from the parental cystatine gene [16]. Exon 10 contains the kinin sequence common for the two kininogens (exon 10a) and a special C-terminal sequence of HMK (exon 10b); exon 11 encodes the unique C-terminal sequence of LMK. The alternative splicing of the primary transcription of the kininogen gene results in two different mRNA specific for HMK and LMK, respectively [17]. Kininogens are polyfunctional glycoproteins consisting of a single polypeptide chain; they are mainly synthesized by hepatocytes and are post-translationally glycosylated and released into the blood flow [17]. The HMK molecule consists of 626 amino acid residues; its molecular weight is ~120 kD and its pI is 4.3. The HMK concentration in human blood plasma is 65-130 µg/ml [18, 19].
The domain structure of HMK is shown in Fig. 1. The heavy chain of HMK includes the first four domains. The D1 domain contains a Ca2+-binding site, and the domain function is not clear in detail [20]. The D2 and D3 domains contain the highly conservative pentapeptide QVVAG, which is a functional inhibitor of cysteine proteinases, such as papain, cathepsins B and H (D2 and D3), and calpain (D2) [21-23].
HMK can reversibly bind to thrombocytes [24, 25], neutrophils [25], and endothelial cells [25, 26], to sites located in the heavy (D3) and light (D5) chains [26] that has been shown by the methods of electron microscopy, monoclonal antibodies to certain antigenic determinants of the HMK molecule, and mutant molecules and their fragments. For the HMK binding to cells, Zn2+ is required. The HMK binding affects the cell functions. Thus, the HMK (D3) interaction with thrombocytes most likely through thrombospondin decreases the activity of thrombocytic calpain, the binding of thrombin, and as a result, inhibits the thrombocyte aggregation stimulated by thrombin [27]. During the binding with multinuclear leukocytes (MNL), HMK acts as an anti-adhesive molecule [27-29]. This effect of HMK seems to be due to the displacement of fibrinogen bound to alpha-, beta2-, and Mac-1-integrins [27, 29, 30]. HMK is also required for the kallikrein-stimulated activation of MNL [30]. And HMK provides an increase in the generation of oxygen free radicals, the degranulation of MNL, and the releasing of granulocytic elastase and lactoferrin [24].Fig. 1. Scheme of the domain structure of high-molecular-weight kininogen of human blood plasma (after [54] with changes). The amino acid sequence 1-18 is the leading peptide. The A-J letters indicate the intron-exon junction. The letters D designate domains. D1 (1-113) is encoded by exons 1, 2, and 3; D2 (114-234) by exons 4, 5, and 6; D3 (235-327) by exons 7, 8, and 9; D4 (358-383) by exon 10 HMK; D5 (384-502) by site 5 of exon 10 HMK; D6 (503-626) by site 3´of exon 10 HMK. The arrows indicate the bonds split by plasma kallikrein. The squares show the location of the carbohydrate chains. The supposed binding sites of cysteine proteinases (D2, D3), thrombospondin (D3), the location of bradykinin (D4), interaction sites with the surface (D5), and binding of factor XI and prekallikrein (D6) are shown by thick lines. Thin lines show disulfide bonds.
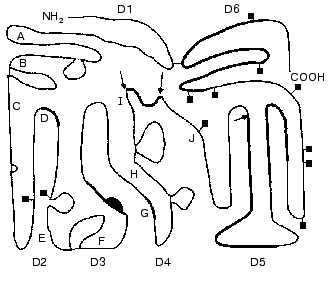
HMK is suggested to bind to endothelial cells through glycoproteins identical to receptor binding the globular regions of the complement C1q-component [31]. Colman [32] has shown that HMK interacts with the receptor of the urokinase-like plasminogen activator on the surface of endothelial cells. On the HMK binding to endothelial cells, the rate of bradykinin release is increased, and the latter stimulates the generation of NO and prostacyclin, which display antithrombotic and vasodilating effects [33].
The bradykinin sequence is located in D4 of kininogen in the disulfide loop flanked by two processing sites for kallikrein-like enzymes. Located in the C-terminal region, the light chain of HMK (D5 and D6, 255 amino acid residues) displays procoagulant activity interacting with hydrophilic and anionic surfaces. The binding to the surface occurs through a D5 region enriched with His, Lys, and Gly residues [34-36], and HMK is involved in formation of a contact ensemble of proteins which is responsible for activation of plasma proteolytic systems as a nonenzymatic cofactor.
The terminal domain of HMK (D6) has sites for binding prekallikrein and the hemocoagulation factor XI.
Thus, the specific features of the domain structure of HMK determine its role in the regulation of functions of a number of blood plasma proteins and of various cells. HMK is involved in the activation of the contact phase of plasma proteolysis, controls the adhesion and activity of thrombocytes, MNL, and endothelial cells, inhibits the activity of cysteine proteinases preventing plasma protein degradation on damage of various tissues, is involved in the regulation of arterial pressure, modifies the inflammatory and antithrombotic reactions, and provides an important regulatory system of interaction between the plasma proteins and blood and vascular wall cells.
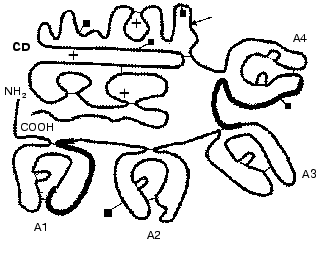
Prekallikrein and kallikrein of blood plasma. Prekallikrein (PK), a precursor of blood plasma kallikrein, is a glycoprotein consisting of 619 amino acid residues. The PK concentration in the blood plasma is 295-580 nM (35-50 µg/ml) [37]. PK is synthesized in hepatocytes. The gene encoding PK consists of fifteen exons and fourteen introns and is located in the distal region of the fourth chromosome [38].
The domain structure of plasma PK is shown in Fig. 2. PK is activated by splitting of the Arg371-Ile372 bond by active forms of blood coagulation factor XII (alphaXIIa and betaXIIa) [15] associated with generation of the light and heavy kallikrein chains bound by a disulfide bond [39]. The light chain contains the catalytic domain typical for serine proteinases with a classic triad of the Ser, His, and Asp residues in the active site of the enzyme. The heavy chain consists of four repeated domains homologous to domains of hemocoagulation factor XI. The heavy chain is involved in the binding of HMK, factor XII, and neutrophils. Colman et al. [40] have shown by inhibitory analysis that PK has two regions interacting with HMK. HMK and PK are bound at 1 : 1 ratio. The HMK sequence of 31 amino acid residues of the D6 domain binds to the N-terminal region of the A1 domain of PK, whereas the C-terminal region binds to the A4 domain of PK. In Figs. 1 and 2, the regions of the binding are shown by thick lines.
Kallikrein resulting on PK activation has a wide spectrum of bioregulatory functions that will be further considered in detail. Kallikrein hydrolyzes two peptide bonds in HMK that releases bradykinin, which in turn controls a great many physiological and pathogenetic processes including the regulation of endothelial cell functions.Fig. 2. Scheme of the domain structure of prekallikrein of human blood plasma (after [54] with changes). A1, A2, A3, and A4 are four apple-shaped domains; CD is the catalytic domain. The arrow points the bond split by active forms of hemocoagulation factor XII on the activation of prekallikrein. The asterisks indicate the catalytic triad specific for serine proteinases (His415, Asp464, and Ser559). The Asp residues (108, 289, 377, 434, and 475) marked by squares are bound to carbohydrate chains. The binding sites for HMK in domains A1 and A4 are shown by thick lines. Thin lines show disulfide bonds.
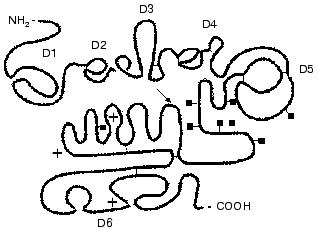
Factor XII of blood coagulation. The domain structure of factor XII is shown in Fig. 3. Factor XII is synthesized in the liver as a single-chain 78-kD glycoprotein containing 7% carbohydrate [41-43]. The mRNA contains ~2100 nucleotides and encodes the sequence of 596 amino acids of the mature protein.
Factor XII consists of several domains: the fibronectin type II domain, the growth factor domain, the fibronectin type I domain, the second growth domain, the kringle domain, and the catalytic domain which is typical for serine proteinases [30, 35, 36, 42, 44, 45]. Factor XII is activated by kallikrein, and HMK increases the reaction rate [46]. This results in a successive generation of two active forms of the factor: alphaXIIa and betaXIIa. The splitting of the Arg353-Val354 bond in the precursor molecule results in the generation of the alphaXIIa form of the enzyme which consists of a heavy and a light chain (353 and 243 amino acid residues, respectively) bound by a disulfide bond [47]. The heavy chain is responsible for the protein binding to the anionic surface upon contact activation. The light chain contains the catalytic domain with amino acid sequence and disulfide bond locations highly homologous to the catalytic domains of plasmin and the tissue- and urokinase-like plasminogen activators (tPA and uPA, respectively) [44]. The active site of factor XIIa forms a classic triad of the His40, Asp89, and Ser191 residues. The betaXIIa form is generated after hydrolysis of two more peptide bonds that results in the generation of a 30-kD enzyme, which is the light chain of the enzyme supplemented with a small fragment of the heavy chain [48, 49]. The betaXIIa and the alphaXIIa forms are more effective in the activation of PK or of factor XI, respectively [49].Fig. 3. Scheme of the domain structure of the factor XII of human blood plasma (after [55] with changes). The arrow shows the Val352-Arg353 bond splitted by kallikrein on the factor XII activation. The light chain contains the catalytic domain (D6), the amino acid residues of the classic triad Asp40, Cys89, and Ser191 are designated by asterisks. The heavy chain contains domains D1 and D3 similar to fibronectin types II and I, domains D2 and D4 (growth factors), and D5 (the kringle domain). The junction places of the carbohydrate chains are marked by squares. Thin lines show disulfide bonds.
Hemocoagulation factor XI. Factor XI is synthesized in the liver as a glycoprotein with molecular weight 143 kD, which contains 5% carbohydrate. It is secreted in the blood plasma as a zymogen at the concentration of ~30 nM. It circulates in a complex with HMK [50]. Factor XI is an unusual zymogen in the serine proteinase family because its molecule consists of two identical peptide chains bound by disulfide bonds [50, 51] and contains two active sites per mole of the enzyme [51].
The factor XI gene is located in the fourth chromosome (4q35); it contains fifteen exons with fourteen introns. The mRNA consists of 2097 nucleotides [52] encoding the protein consisting of 607 amino acid residues. Each of two chains in the mature molecule consists of two heavy chains each of 369 amino acids and of two light chains each of 238 amino acids, which are kept together by disulfide bonds. Each heavy chain of factor XI has four repeated domains with 23-34% homology [52]. Each domain contains 90-91 amino acid residues and three disulfide bonds. These domains and also the catalytic domain of factor XI are highly homologous to the corresponding domains of PK. The repeated domains inherent in PK and in factor XI are not found in any other protein. Each light chain also includes the typical N-terminal sequence (Ile-Val-Gly-Gly-) and the catalytic triad of His44, Asp93, and Ser188, which is specific for most serine proteinases. Factor XI has five sites for glycosylation by amino groups of Asp72, Asp108, and Asp335 of the heavy chain and of Asp63 and Asp104 of the light chain [53].
Factor XI is activated by factor XIIa in the presence of HMK due to hydrolysis of the Arg369-Ile370 bond in each polypeptide chain.
To finish the description of properties of the contact system components, attention should be paid to a fundamental concept: the proteins of this system are not essential elements of the hemocoagulating pathway but only play an auxiliary role of secondary importance in the generation of thrombin. Thus, the deficiency of factor XII, PK, and HMK does not result in a pronounced hemophilia.
The activation of the contact system and the mechanism of PK interaction, hemocoagulation factors XII and XI, and HMK, were studied in vitro in detail on an activating polyanionic surface [54-57, 59, 60].
At present, the conception of the mechanism of activation and the role of the contact system in the regulation of hemostasis is under significant revision. Earlier this system was mainly considered as an initial stage of the internal pathway of the hemocoagulating cascade, but now it has become clear that this system initiates fibrinolysis. Kallikrein (and to a smaller degree factors XIIa and XIa) directly activates plasminogen, although less effectively than uPA. Nevertheless, plasma kallikrein is already characterized as a kinetically effective in vitro activator of pro-uPA [61]. Activation of pro-uPA was found on the surface of thrombocytes and endothelial cells [62-64]. Pro-uPA is most effectively activated on kallikrein binding through HMK to the uPA receptor [31]. Moreover, the high homology in the structure of factor XII, plasminogen, and tissue plasminogen activators suggests that the contact system is mainly a system of fibrinolysis [49]. Extensive experimental and clinical studies have shown that a contact system is required for initiation, enhancement, and propagation of protective reactions, which are provided by these systems and by other systems interacting with them. The enzymes of the contact system activate plasminogen, the first component (C1) and factor B of the complement, hemocoagulation factor VII, and prorenin, and also stimulate the activation of neutrophils and influence endothelial cell functions directly or through the releasing of bradykinin. Bradykinin is known to play a main role in the development of inflammation [65]. Especially important is the bradykinin property of releasing cytokines, such as interleukin 1, tumor necrosis factor, and other secondarily generated mediators: NO, prostaglandins, and leukotrienes [42].
The hypothesis of the contact activation of factor XII and PK on binding with a negatively charged surface is the basis for widely used coagulational tests, such as the activated partial thromboplastin time, and also for concepts on the possible in vivo activation of KKS on artificial surfaces on medical interventions and infection of the body.
Negatively charged physiological surfaces such as sulfatides, phospholipids, urates, cholesterol and chondroitin sulfates, and heparins and other glycosamines can be present and appear at various damages of the body. They can be theoretically considered as physiological anionic surfaces, but there is no convincing evidence. Therefore, the question of the contact system role in vivo remains open.
Moreover, a careful analysis of properties of all components of the internal and external pathways of activation of the hemocoagulating cascade including their involvement in the biological reactions in this pathway and in the development of pathological processes associated with hemophilia manifestations has shown that the deficiency of factor XII, PK, and HMK does not result in bleeding in patients, although the time of the in vitro blood coagulation is markedly prolonged. The activation of factor XI by thrombin without involvement of the contact system that was found in the early 90s [66] also suggests that the contact system has functions beyond hemocoagulation.
It should be noted that notwithstanding the significantly enlarged knowledge of the role of the contact system in the activation of the plasma proteolytic systems and functions of blood cells, this system is traditionally considered in connection with the activation of the internal pathway of blood coagulation. This standpoint was appropriated as a paradigm and existed during the 35-year history of studies on the contact phase. However, at present, many clinical and experimental data are accumulated suggesting that the main physiological role of this system should be beyond hemocoagulation. This is also supported by the recent literature data, which suggest an important involvement of the contact system (including the kallikrein-kinin system) in vascular biology. In particular, this relates to its role in the maintaining of vascular wholeness and blood pressure, in the influence on various functions of endothelial cells, in the control of fibrinolysis, and in maintaining of the constitutive anticoagulant character of the intravascular space [67-71].
ACTIVATION OF THE CONTACT SYSTEM ON THE ENDOTHELIAL CELL
SURFACE
From the middle of the 90s, the hypothesis of the contact activation on a negatively charged surface has been under careful testing, and this has resulted in its revision [67]. A new concept on the interaction and activation of the contact system components on cell membranes was proposed which explained the in vivo role of this system of proteins. Studies of Colman and Schmaier, et al. [67, 70-72] made essential contributions to understanding of the contact system activation on endothelial cells. Their hypothesis is alternative to the autoactivating mechanism of factor XIIa generation accompanied by the activation of the kallikrein-kinin system proteins. Their hypothesis is based on the idea that the in vivo assembly of multicomplexes of the contact system components occurs on cell receptors. For 10 years receptors to the contact system proteins were searched for on cell membranes and the composition of the protein ensemble was studied as the place of the PK and factor XII activation [62, 67-75].
The concept proposed suggests that the contact system is activated on the surface of endothelial cells [71, 72]. Studies on the HMK-PK complex binding to the cell membrane proteins have shown that, notwithstanding the interaction of factor XII with this complex, PK is activated on endothelial cells does not depend on the presence of factor XII or its active forms [76, 77].
The binding of HMK to the endothelial cell membranes was found to be controlled by protein kinase C and by the bradykinin B1 receptor expressed on the cell surface. Inhibitors of the angiotensin-converting enzyme increase this effect of bradykinin, whereas HMK more effectively binds to the membrane [74, 75, 78]. Because the light chain of HMK is complexed with PK and XI, these components are bound to endothelial cells through HMK. The activation of PK and the generation of kallikrein initiate the generation of bradykinin, which in its turn enhances the binding of HMK to the cells.
By affinity chromatography from lysates of endothelial cells, the main protein capable of binding to HMK was isolated and identified as cytokeratin 1 (CK1), a typical protein of interstitial fibers of the cytoskeleton. The exposition of this protein on the endothelial cell membrane was also shown by laser scanning microscopy and flow cytometry [74, 78]. HMK specifically interacts with native or recombinant CK1 only in the presence of Zn2+ [74]. The HMK-binding site of CK1 is located in a protein segment consisting of 20 amino acids. CK1 was also found on thrombocytes and granulocytes. Finally, the peptides covering the binding site of CK1 inhibit the activation of PK on endothelial cells, which is supported by involvement of CK1 in the ensemble required for the activation of PK [75]. In addition to CK1, the uPA receptor (uPAR) and the receptor of the q-subunit of the complement first component (gC1qR) are involved in formation of the complex, which binds HMK by endothelial cells and granulocytes. Similarly to CK1, these proteins interact with the immobilized HMK on affinity chromatography [74, 78].
Studies on the inhibitory spectrum and substrate specificity of the endothelium surface-bound proteinase, which is required to activate PK, resulted in its determination as a Zn2+-dependent cysteine proteinase [74, 78]. But it is not a typical cysteine proteinase because it is not suppressed by iodoacetate or by N-ethylmaleimide. This proteinase is not calpain because its activity is not suppressed by the inhibitor E64. Finally, inhibitors of metalloproteinases TIMP-1, TIMP-2, or BB94 fail to inhibit the activation of PK. By data of Schmaier and Rojkjaer [69], this unique proteinase, which activates PK, is not associated with the protein ensemble binding HMK and PK to cells. PK is activated by an antipain-sensitive membrane proteinase in the case of interaction of the protein ensemble with a large number of microvascular endothelium cells. The authors suggest that such mechanism of PK activation can be fundamental for many cells during the binding of the PK-HMK complex to their membranes. Studies on the activation kinetics of PK and factor XII have shown that the activation of PK is the first stage of physiological activation of KKS on endothelial cells. Factor XII is activated after the activation of PK under the influence of kallikrein.
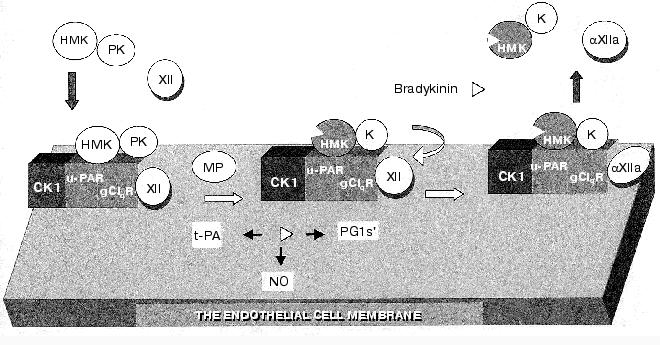
The model of the main events in KKS activation on the endothelial cell is presented in Fig. 4. Conditions favorable for the KKS activation are provided by generation on the cell surface of a polyprotein ensemble, which, in addition to HMK and PK, also includes the uPA receptor (uPAR), the receptor of the q-subunit of the complement first component (gC1qR), factor XII, and cytokeratin 1 [78-81]. In this complex PK is activated under the influence of the membrane-bound cysteine proteinase (MP) with generation of kallikreins, which in turn activates factor XII. Kallikrein splits HMK; this is associated with generation of bradykinin, and the complex of dekininated HMK with kallikrein is removed from the endothelial cell surface. This new hypothesis on the kallikrein-kinin system activation suggests its important role in the regulation of biological functions of the vascular wall [82].
Because the factor XI structure is very like that of PK, its activation mechanism is virtually identical to that of PK and needs the interaction with HMK on endothelial cells in the presence of Zn2+ and of an antipain-sensitive proteinase. This mechanism of factor XI activation with involvement of the above-described protein ensemble on endothelial cells is independent of factor XIIa, autoactivation, and alpha-thrombin [67, 69]. At present, it is unclear how this mechanism of activation of factors XI and XII is involved in hemostasis.Fig. 4. Model of the KKS activation on endothelial cells (after [76] with changes). For explanations, see the text. Designations: PG1s´. prostaglandin 1s´; CK1, cytokeratin 1; K, kallikrein.
However, these studies convincingly show the important role of PK and of the kallikrein-kinin system as a whole in the cell membrane biology. This especially refers to bradykinin [20, 22, 69]. Bradykinin is a physiological and the strongest in vivo stimulator of tPA release, and this suggests its important contribution to fibrinolysis. It is reasonable to consider bradykinin generation as the crucial event in the regulation of arterial pressure and modulation of many other processes determining vascular functions. Kallikrein is a kinetically favorable activator of pro-uPA [64], and fibrinolysis can be activated without the involvement of factor XII. It should be remembered that HMK, in particular, inhibits the thrombin-initiated activation of thrombocytes. First of all, this is associated with the inhibition by kininogen of the calpain-induced aggregation of thrombocytes. Calpain is expressed on the membrane and initiates the complexing of integrins alphaIIbbeta3, which are required for fibrinogen binding and thrombocyte aggregation. Moreover, kininogens suppress the binding of alpha-thrombin to thrombocytes and endothelial cells.
In addition to the function of kininogenase and the involvement in the regulation of activities of other proteolytic systems, blood plasma kallikrein also stimulates the activation of MNL associated with elastase release from azurophilic granules and with activation of the latent collagenase [83]. All these properties of blood plasma kallikrein and also the bioregulatory functions of bradykinin and HMK are responsible for the involvement of the KKS in various important processes which under certain conditions can provoke some pathological states. Thus, clinical and experimental studies show KKS involvement in the pathogenesis of acute and chronic inflammation, shock of different etiologies, diabetes, allergy, thrombo-hemorrhagic disorders including disseminated intravascular blood coagulation, oncological diseases, and many other pathological states of the body [37, 42, 58, 59, 65, 84].
Pathogenetic functions of the blood plasma kallikrein are mainly displayed under shortage of its inhibitors. The kallikrein activity in the blood plasma is mainly controlled by the inactivator of the complement first component (IC1), alpha2-macroglobulin, and to a lesser degree by antithrombin III and the inhibitor of protein C [85, 86].
INTERACTION OF PROTEINS OF THE BLOOD PLASMA CONTACT SYSTEM WITH
SOME MULTINUCLEAR LEUKOCYTIC PROTEINASES DURING INFLAMMATION
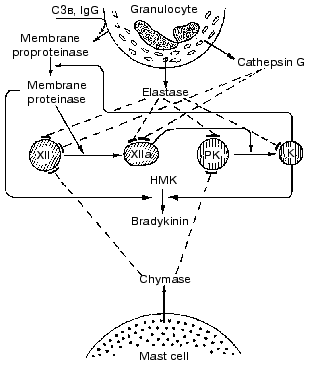
There are diverse complicated interactions of contact phase components with blood cell proteinases in the development of inflammation. In particular, depending on the stage of the MNL activation, these cells can activate or inactivate the contact system proteins [87]. We studied the effect of MNL on highly purified preparations of factors XII and XIIa, prekallikrein, and kallikrein isolated from blood plasma and found that at the initial activation stage (priming) MNL could release an earlier unknown serine proteinase that after activation by kallikrein dose-dependently activated factor XII. However, during the secretory degranulation of these cells accompanied by release of lysosomal proteinases from the azurophilic granules the contact system components are inactivated. Purified preparations of elastase and cathepsin G isolated from the leukocytes of donors' blood inactivated preparations of kallikrein, factor XIIa, and their zymogens [87]. Chymase of mast cells also inactivated zymogens [87]. Our data generalizing the possible mechanisms of interaction of key proteinases of the contact system and of proteinases of MNL and mast cells are presented in Fig. 5.
Our hypothesis on the interaction between MNL and contact system components is supported by many clinical observations [87-92] of an increase in the activation of PK depending on inflammation severity and of a subsequent dramatic decrease in kallikrein activity and in the level of its precursor along with a steady increase in the activity of elastase in the terminal stage of disease, especially in cases with unfavorable outcome [87-89].Fig. 5. Scheme of interaction between proteins of the blood plasma contact system and certain proteinases of granulocytes and mast cells. The arrows designate the activation and conversion and the dashed lines show the inactivation. K, kallikreins.
This hypothesis, in particular, is supported by the following experiment: purified leukocytic elastase was added into donors' blood plasma in quantities corresponding to the amount of elastase released in the blood on MNL degranulation during inflammation, and then factor XIIa, kallikrein, and their precursors were determined. The findings were compared to determinations of these components in the blood plasma of patients with peritonitis at the critical state.
The contact system components were assessed by our method of concurrent determinations of factors XII, XIIa, PK, and kallikrein using two chromogenic substrates, Chromozym PK (Bz-Pro-Phe-Arg-pNA) and S-2302 (H-D-Pro-Phe-Arg-pNA 2HCl) [93]. The essence of the method is the determination of the total amidase activity of factor XIIa and kallikrein in the blood plasma by the rate of hydrolysis of S-2302 and of the kallikrein activity by the rate of hydrolysis of Chromozym PK. The difference of these activities allows us to determine factor XIIa. To determine the precursors, they are activated by dextran sulfate and the enzyme activities are determined as described.
The activity of elastase was determined after the enzyme liberation from the complex with alpha1-PI by our method described in [94]. This method was compared to enzyme immunoassay by which elastase can be determined in the complex with alpha1-PI [89, 92]. These determinations were performed in parallel at Munich University and at the Russian Medical Academy of Postgraduate Education. The blood plasma of patients with multiple traumas was used as obtained in the Surgery Clinic, Munich University. The methods were comparable in the prediction of complications (sepsis) and of termination of the critical state (unpublished).
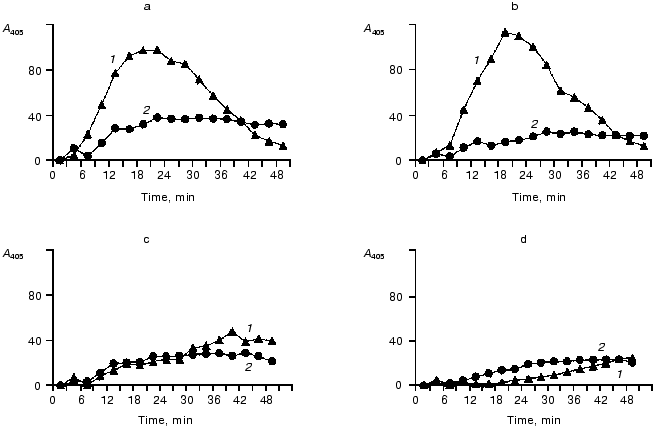
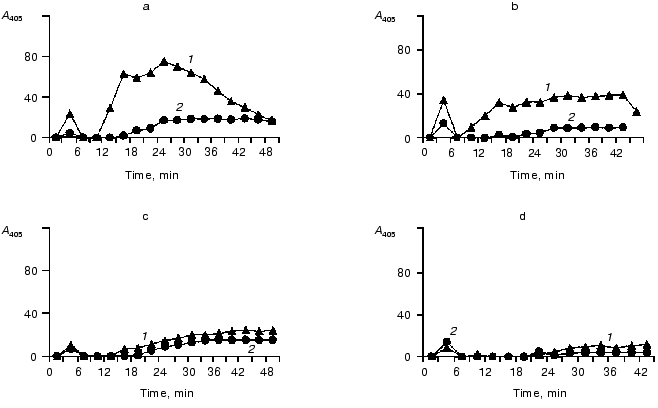
Effects of elastase on the contact system components in the blood plasma of donors and of patients with peritonitis are presented in Figs. 6 and 7, which show a similarity of the activity curves of the contact system components studied. These findings suggest that during an active inflammation the system components can be damaged by elastase that in turn can result in exhaustion and disorders of their protective functions.
Fig. 6. Activities of kallikrein, factor XIIa, and their zymogens depending on the measurement time in donor's blood plasma before or after the addition of leukocytic elastase. The decrease in the proteinase activities after the maximum is explained by the effects of inhibitors of these enzymes; a) donor's blood plasma; b, c, d) donor's blood plasma after the addition of 18.7, 50, 62.5 mU of elastase (by hydrolysis of Meo-Suc-Ala-Ala-Pro-Val-pNA); 1) S-2302; 2) Chromozym PK.
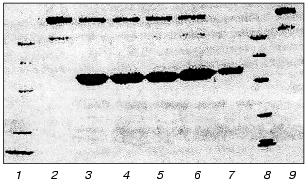
We have studied the effect of leukocytic elastase on HMK together with H. Johansen (Department of Pharmacology, Oslo University) [95]. Incubation of purified HMK preparations with leukocytic elastase resulted in the splitting of HMK molecules. Figure 8 presents an electrophoregram of HMK and products of its degradation after its incubation with elastase for 20 sec and 1, 2, 3, and 4 min. HMK in the sample incubated was split at the rate of 467 µmole/min per mg HMK.Fig. 7. Activities of kallikrein and of factor XIIa and contents of their zymogens in blood plasma of a patient with peritonitis (before the operation and at different times after it). The elastase activity per mg protein of the blood plasma before the operation was 12 mU (a), on the third (b), fourth (c), and seventh (d) after the operation it was 44, 44, and 69 mU, respectively; 1) S-2302; 2) Chromozym PK.
HMK was incubated with the leukocytic elastase complex with alpha1-PI, and, notwithstanding the complete inhibition of amidase activity of the enzyme, it retained the ability to split HMK taken in equimolar and two-, four-, and fivefold higher concentrations. The electrophoregram of products resulting on the long-term incubation of HMK with the earlier prepared leukocytic elastase complex with alpha1-PI is presented in Fig. 9.Fig. 8. Effect of leukocytic elastase on high-molecular-weight kininogen of human blood plasma. SDS-electrophoregram in 10% polyacrylamide gel, staining with Coomassie R-250: 1) HMK; 2-6) HMK + elastase (1 (2), 2 (3), 3 (4), 4 (5), and 5 min (6)); 7) elastase; 8) set of standard proteins. The incubation mixture contained 200 µl of HMK (0.55 nM), 10 µl of elastase (34.5 nM), and 30 µl of 0.1 M Tris-HCl buffer (pH 8.6). After the incubation aliquots of 30 µl were supplemented with 10 µl of 8% SDS and kept in a boiling bath for 5 min.

Kininogen was degraded notwithstanding the presence of alpha1-PI that was confirmed by immunoelectrophoretic determination of kininogen in the citrated plasma after the addition of elastase. The cause of this unexpected hydrolytic effect of the leukocytic elastase on high-molecular-weight kininogen of the human blood plasma notwithstanding the presence of alpha1-PI is now under study.Fig. 9. Results of the HMK incubation with the complex of leukocytic elastase (LE) with alpha1-PI. SDS-electrophoregram in 10% polyacrylamide gel, staining with Coomassie R-250. The molar ratio LE/alpha1-PI in the complex is 1 : 2. The incubation mixture contained 200 µl of HMK and 40 µl of the LE-alpha1-PI complex; 1, 8) set of standard proteins; 2) HMK; 3) HMK + LE-alpha1-PI (20 sec); 4) HMK + LE-alpha1-PI (1 h); 5) HMK + LE-alpha1-PI (3 h); 6) HMK + LE-alpha1-PI (20 h); 7) LE-alpha1-PI; 9) HMK.
Our findings and the literature data suggest that not only the contact system factors but also other blood plasma proteins are split in vivo under the influence of elastase. This finding can contribute to the understanding of the pathogenetic role of leukocytic elastase in the development of thrombo-hemorrhagic complications during severe inflammations, such as multiple trauma, peritonitis, and septicemia [92-96]. The uncontrolled effect of elastase on the contact system proteins was the highest under conditions of hereditary or acquired deficiency of alpha1-PI and other inhibitors of the system proteinases that is often observed in critical states with unfavorable termination [92-96].
In studies on the so-called contact system the composition of this protein ensemble was established, the protein structures and functional properties were described, and the protein-protein interactions were presented in detail. Earlier the contact system was thought to be activated on a polyanionic surface, and factor XII autoactivation seemed the most likely triggering mechanism. Now the HMK ensemble on the polyprotein receptor has been shown to be formed on endothelial cells and to result in activation of PK. Factor XII is activated secondarily by the kallikrein produced, and then the process is amplified due to reciprocal activation of PK and factor XII. Factor XI also bound in the protein ensemble on endothelial cells can be activated by the same mechanism as PK. Kallikrein concurrently splits HMK with generation of bradykinin, which stimulates the release of the tissue activator plasminogen. Moreover, the kallikrein-kinin system components seem to be involved in the processing of urokinase. And, finally, HMK and products of its hydrolysis display direct and mediated antithrombin and anticoagulant effects.
The structures and functions of the contact system proteins can be damaged by proteinases released by blood cells in diseases. This conclusion is based on the finding of the destructuring effect of elastase and cathepsin G on HMK, factors XII, XIIa, and PK, and kallikrein also confirmed by clinical studies.
REFERENCES
1.Wachtfogel, Y. T., de la Cadena, R. A., and Colman,
R. W. (1993) Thromb. Res., 72,1-21.
2.Mitropoulos, K. A. (1999) Thromb. Res.,
94,117-129.
3.Griep, M. A., Fujikawa, K., and Nelsestuen, G. L.
(1985) Biochemistry, 24,4124-4130.
4.Tans, G., Rosing, J., and Griffin, J. H. (1983)
J. Biol. Chem., 258,8215-8222.
5.Etspana, F., and Ratnoff, O. D. (1983) J. Lab.
Clin. Med., 102,31-45.
6.Tankersley, D. L., and Finlayson, J. S. (1984)
Biochemistry, 23, 273-279.
7.Silverberg, M., Dunn, J. T., Garen, L., and Kaplan,
A. P. (1980) J. Biol. Chem., 255,7281-7286.
8.Dunn, J. T., Silverberg, M., and Kaplan, A. P.
(1982) J. Biol. Chem., 257, 1779-1784.
9.Mandle, R., Jr., Colman, R. W., and Kaplan, A. P.
(1976) Proc. Natl. Acad. Sci. USA,73,4179-4183.
10.Cochrane, C. G., and Revak, S. D. (1980) J.
Exp. Med., 152, 608-619.
11.Miles, L. A., Greengard, J. S., and Griffin, J.
H. (1983) Thromb. Res., 9, 407-417.
12.Ghebrehiwet, B., Silverberg, M., and Kaplan, A.
P. (1981) J. Exp. Med., 153, 665-676.
13.Ikari, N., Niinobe, M., and Fujii, S. (1982)
Agents Actions (Suppl.), 9, 35-39.
14.Colman, R. W. (1984) J. Clin.
Invest.,73, 1249-1253.
15.Goldsmith, G. H. (1980) J. Lab. Clin.
Med., 96, 564-566.
16.Kitamura, N., Kitagawa, H., Fukushima, D.,
Takagaki, Y., Miyata, T., and Nakanishi, S. (1985) J. Biol.
Chem., 260,8610-8617.
17.Takagaki, Y., Kitamura, N., and Nakanishi, S.
(1985) J. Biol. Chem., 260, 8601-8609.
18.Hoem, N.-O., Johansen, H. T., Johannesen, S., and
Briseid, K. (1989) Adv. Exp. Med. Biol., 247A,
337-343.
19.Adam, A., Albert, A., Calay, G., Closset, J.,
Damas, J., and Franchimont, P. (1985) Clin. Chem.,
31,423-426.
20.Higashiyama, S., Ohkubo, I., Ishiguro, H.,
Sasaki, M., Matsuda, T., and Nakamura, R. (1987) Biochemistry,
26,7450-7458.
21.Barret, A. J., Rawlings, N. D., Davies, M. E.,
Machleidt, W., Salvesen, G., and Turk, V. (1986) J. Biol.
Chem., 260,8610-8617.
22.Müeller-Esterl, W., Fritz, H., Machleidt,
W., Ritonjga, A., Brzin, J., Kotnik, M., Turk, V., Kellermann, J., and
Lottspeich, F. (1985) FEBS Lett., 182, 310-314.
23.Bradford, H. N., Jameson, B. A., Adam, A. A.,
Wassell, R. P., and Colman, R. W. (1993) J. Biol. Chem.,
268,26546-26551.
24.Gustafson, E. J., Schutsky, D., Knight, L. C.,
and Schmaier, A. H. (1986) J. Clin. Invest., 78,
310-318.
25.Van Iwaarden, F., de Groot, P. G., Sixma, J. J.,
Berrettini, M., and Bouma, B. M. (1988) Blood, 71,
1268-1276.
26.Herwald, H., Hasan, A. A. K., Godovac-Zimmermann,
J., Schmaier, A. H., and Müller-Esterl, W. (1995) J. Biol.
Chem., 270, 14634-14642.
27.Gustafson, E., Lukasiewicz, H., Wachtvogel, Y.,
Norton, K., Schmaier, A., Niewarowski, S., and Colman, R. (1989) J.
Cell Biol., 109,377-387.
28.Colman, R. W. (1990) Adv. Exp. Med. Biol.,
281, 195-200.
29.Wachtvogel, Y. T., de la Cadena, R. A., Kunapuli,
S. P., Rick, L., Miller, M., Schultze, R. L., Altieri, D. C.,
Edgington, T. S., and Colman, R. W. (1994) J. Biol. Chem.,
269, 19307-19312.
30.Colman, R. W. (1996) Immunopharmacology,
32, 9-18.
31.Herwald, H., Dedio, J., Kellner, R., Loos, M.,
and Müeller-Esterl, W. (1996) J. Biol. Chem., 271,
13040-13047.
32.Colman, R. W. (1996) Circulation,
94, 1-42.
33.D'Orleans-Juste, P., de Nucci, G., and Vane, J.
P. (1989) Br. J. Pharmacol., 96, 920-926.
34.Kunapuli, S. P., de la Cadena, R. A., and Colman,
R. W. (1993) J. Biol. Chem., 268, 2486-2492.
35.Schmaier, A. H., Schutsky, D., Farber, A.,
Silver, L. D., Bradford, H. N., and Colman, R. W. (1987) J. Biol.
Chem., 262, 1405-1411.
36.De la Cadena, R., and Colman, R. (1992)
Protein Sci., 1, 151-160.
37.Schmaier, A. H., Silverberg, M., Kaplan, A. P.,
and Colman, R. W. (1987) Hemost. Thromb., 18-37.
38.Beaubien, G., Rosinski-Chupin, I., Mattei, M. G.,
Mbikay, M., Chretien, M., and Siedan, N. G. (1991) Biochemistry,
30, 1628-1635.
39.Page, J. D., and Colman, R. W. (1991) J. Biol.
Chem., 266, 8143-8148.
40.Page, J. D., Yen, J. L., Harris, R. B., and
Colman, R. W. (1994) Arch. Biochem. Biophys., 314,
159-164.
41.Que, B. G., and Davie, E. W. (1986)
Biochemistry,25, 1525.
42.Bhoola, K. D., Figueroa, C. D., and Worthy, K.
(1992) Pharmacol. Rev., 44, 1-60.
43.Scott, C. F., Silver, L. D., Schapira, M., and
Colman, R. W. (1984) J. Clin. Invest., 73, 954-962.
44.Fujikawa, K., and McMullen, B. A. (1983) J.
Biol. Chem., 258,10924.
45.McMullen, B. A., and Fujikawa, K. (1985) J.
Biol. Chem., 260, 5328.
46.Fujikawa, K., Heimark, R. L., Kurachi, K., and
Davie, E. W. (1980) Biochemistry, 19, 1322.
47.Fujikawa, K., and Davie, E. W. (1981) Meth.
Enzymol., 80, 198-211.
48.Dunn, J. T., and Kaplan, A. P. (1982) J. Clin.
Invest., 70, 627-631.
49.Tans, G., and Rosing, J. (1987) Sem. Thromb.
Hemost., 13, 25-35.
50.Thsby, S., Kumar, A., and Joseph, M. (1985)
Nature, 316, 271.
51.Kurachi, K., and Davie, E. W. (1977)
Biochemistry,16, 5831.
52.Fujikawa, K., Chung, D. W., Hendrickson, L., and
Dane, E. W. (1986) Biochemistry,25,2417.
53.Earl, W., and Davie, E. (1987) Thromb.
Hemorrhage, 242-267.
54.Colman, R. W. (1996) Immunopharmacology,
32, 9-18.
55.Davie, E. W., Fujikawa, K., and Kisiel, W. (1991)
Biochemistry, 30, 10363-10370.
56.Retxios, A. D., Rosenfeld, R., and Schiffman, S.
(1987) J. Biol. Chem., 262, 3074-3081.
57.Tayeh, M. A., Olson, S. T., and Shore, J. D.
(1985) J. Biol. Chem., 260, 10856-10863.
58.De la Cadena, R. A., Wachtvogel, Y. T., and
Colman, R. W. (1974) Hemost. Thromb., 219-240.
59.Stadnicki, A., de la Cadena, R., Sartor, R. B.,
Kettner, C. A., and Colman, R. W. (1996) Immunopharmacology,
33, 314-316.
60.Scott, C. F., Silver, L. D., Purdon, A. D., and
Colman, R. W. (1985) J. Biol. Chem., 260,
10856-10863.
61.Ichinose, A., Fujikawa, K., and Suyama, T. (1986)
J. Biol. Chem., 261, 3486-3489.
62.Lenich, C., Pannell, R., and Gurewich, V. (1995)
Thromb. Haemost., 74, 698-703.
63.Loza, J. P., Gurewich, V., Johnstone, M., and
Pannel, R. (1994) Thromb. Haemost., 71, 347-352.
64.Gurewich, V., Johnstone, M., Loza, J. P., and
Pannel, R. (1993) FEBS Lett., 318, 317-321.
65.Colman, R. W. (1993) Agents Actions
(Suppl.), 42, 125-143.
66.Gailani, D., and Broze, G. J. (1991)
Science,253,909-912.
67.Colman, R. W., and Schmaier, A. H. (1997)
Blood, 90, 3819-3843.
68.Brown, N. J., Nadeau, J. H., and Vaughan, D. E.
(1997) Thromb. Haemost., 77, 522-525.
69.Schmaier, A. H., and Rojkjaer, R. (1999)
Thromb. Haemost.,82, 226-233.
70.Schmaier, A. H., Kuo, A., Lundberg, D., Myrray,
S., and Cines, D. B. (1988) J. Biol. Chem., 263,
16327-16333.
71.Rojkar, R., and Schmaier, A. H. (1999) Proc.
Assoc. Am. Phys., 111, 220-227.
72.Kusuman, Y., Shibayama, Y., and Kaplan, A. P.
(1999) Immunopharmacology, 43, 203-210.
73.Joser, K., Shibayama, Y., Nakazava, Y.,
Peerschke, E. I. B., Ghebrehiwet, B., and Kaplan, A. P. (1999)
Immunopharmacology,43,155-161.
74.Joseph, K., Ghebrehiwet, B., Peerschke, E. I. B.,
Reid, K. B. M., and Kaplan, A. P. (1996) Proc. Natl. Acad. Sci.
USA, 93,8552-8557.
75.Colman, R. W., Pixley, R. A., Najamunnisa, S.,
Yan, W.-Y., Wang, J., Mazar, A., and McCrae, K. R. (1997) J. Clin.
Invest., 100, 1481-1487.
76.Rojkjaer, R., Motta, G., Hasan, A. A. K.,
Schousboe, I., and Schmaier, A. H. (1998) Thromb. Haemost.,
80, 74-81.
77.Colman, R. W., Lin, Y., Johnson, D., and Mousa,
S. A. (1998) Blood,998,92, 174a.
78.Herwald, H., Dedio, J., Kellner, R., Loos, M.,
and Mueller-Esterl, W. (1996) J. Biol. Chem., 273,
1551-1555.
79.Dedio, J., and Mueller-Esterl, W. (1996) FEBS
Lett., 399,255-258.
80.Dedio, J., Jahnen-Dechent, W., Bachmann, M., and
Mueller-Esterl, W. (1998) J. Immunol.,
160,3534-3542.
81.Van den Berg, R. H., Prins, F., Faber-Krol, M.
C., Lynch, N. J., Schwarble, W., van Es, L. A., and Daha, M. R. (1998)
J. Immunol.,158,3909-3916.
82.Zini, J. M., Schmaier, A. H., and Cines, D. B.
(1993) Blood, 81, 2936-2946.
83.Kaplan, A. P., and Silverberg, M. (1987)
Blood, 70, 1-15.
84.Cardin, A. D., Jackson, R. L., Donaldson, V. H.,
Chao, J., and Margolius, H. S. (1986) Adv. Exp. Med. Biol.,
198 (Pt. B), 195-202.
85.Schapira, M. (1987) Sem. Thromb. Hemost.,
13, 69-78.
86.Espana, F., Estelles, A., Griffin, J. H., and
Aznar, J. (1991) Thromb. Haemost., 65, 46-51.
87.Yarovaya, G. A., Dotsenko, V. L., and Neshkova,
H. A. (1991) Agents Actions (Suppl.), 38, 144-156.
88.Yarovaya, G., Shutov, E., Jebelenco, G.,
Dotsenko, V., and Neshkova, E. (1996) Immunopharmacology,
32, 135-140.
89.Fritz, H., Jochum, H., Geiger, R., Duswald, Kh.,
Dittmer, H., Kortmann, H., Neumann, S., and Lang, H. (1986) Filia
Histochem. Cytobiol., 24, 99-119.
90.Kaplan, A. P., and Joseph, K. (1997) Adv.
Immunol., 66, 225-272.
91.Shcherbakova, I., Neshkova, E., Dotsenko, V.,
Platonova, T., Shcherbakova, E., and Yarovaya, G. (1999)
Immunopharmacology, 43, 273-279.
92.Jochum, M., Machleidt, W., and Fritz, H. (1993)
Handbook of Mediators in Septic Shock, CRC Press, London-Tokyo,
pp. 335-361.
93.Blokhina, T. B., Vasil'ev, I. T., Babaev, I. B.,
Neshkova, E. A., Dotsenko, V. L., and Yarovaya, G. A. (2000)
Abst. Conf. on the 90th Anniversary of the Botkin City
Clinical Hospital, Committee of Health, Moscow, pp. 26-27.
94.Dotsenko, V. L., Neshkova, E. A., and Yarovaya,
G. A. (1994) Vopr. Med. Khim., 40, 20-25.
95.Dotsenko, V. L., Neshkova, E. A., Rugness, E.,
Johansen, H., Blokhina, T. B., and Yarovaya, G. A. (2001) Vopr. Med.
Khim., 47, 55-70.
96.Yarovaya, G. A. (2001) Vopr. Med. Khim.,
47, 20-42.