Determination of a Non-methylated Deoxycytidine Residue in the Recognition Site of DNA-methyltransferases
E. A. Kubareva1*, J. Walter2, O. V. Vorob'eva1, M. V. Razumikhin3, A. S. Karyagina3, P. C. K. Lau4, and T. Trautner2
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: kubareva@genebee.msu.su2Max-Planck-Institut für Moleculare Genetic, Ihnestrasse 73, Berlin D-14195, Germany; fax: (49) 30-8413-1382; E-mail: j.walter@mx.uni-saarland.de
3Institute of Agricultural Biotechnology, Timiryazevskaya ul. 42, Moscow, 127550 Russia; fax: (095) 977-0947; E-mail: anna@jab.ac.ru
4Biotechnology Research Institute, National Research Council of Canada, Montreal, Quebec H4P 2R2, Canada; fax: (1-514) 496-6265; E-mail: Peter.Lau@nrc.ca
* To whom correspondence should be addressed.
Received June 8, 2001
A method for determination of a non-methylated deoxycytidine (dC) residue in the recognition site of 5-cytosine DNA-methyltransferases is suggested. The method is based on treatment of methylated DNA by sodium bisulfite and successive reaction of the thus modified DNA with a repair enzyme, uracil-DNA glycosylase. This method was successfully applied to identify NlaX methyltransferase specificity.
KEY WORDS: 5-cytosine DNA-methyltransferase, non-methylated and methylated bases, bisulfite reaction, uracil-DNA glycosylase
Abbreviations: SAM) S-adenosyl-L-methionine; UDG) uracil-DNA glycosylase; M.NlaX) DNA-methyltransferase NlaX; M.SsoII) DNA-methyltransferase SsoII; R.SsoII) restriction endonuclease SsoII.
Some purine and pyrimidine bases can be modified after the end of the
DNA replication cycle. The modified nucleosides,
5-methyl-2´-deoxycytidine, N6-methyl-2´-deoxyadenosine, and
N4-methyl-2´-deoxycytidine, are thus formed in DNA. The methyl
group is incorporated into the bases by site-specific
DNA-methyltransferases in the presence of the cofactor
S-adenosyl-L-methionine (SAM), the donor of the methyl group. DNA
methylation has been shown to be very important for regulation of
expression of many genes and in DNA replication and repair. In
eukaryotic systems, DNA methylation is also significant for such
important processes as genome imprinting and X-chromosome inactivation
[1, 2]. Most bacterial
DNA-methyltransferases are components of restriction-modification
systems, a peculiar kind of immune system protecting bacteria from
infection by bacteriophage. In addition to methyltransferase, these
systems also contain restriction endonuclease that cleaves the phage
DNA penetrating into a cell in a certain place, the recognition site.
The cell DNA is not hydrolyzed by restriction endonuclease because the
former is modified by DNA-methyltransferase within the recognition site
[3].
Biochemical studies of DNA-methyltransferase are impossible without determination of its recognition site in DNA, methylation position and its type. There are several ways of determining the position of the base methylated by DNA-methyltransferases: destruction of the methylated DNA to short oligonucleotides, identification of methylated fragments, and their sequencing [4, 5]; detection of tritium-containing DNA fragments. Such fragments are formed after DNA methylation in the presence of SAM having tritium-labeled methyl group and subsequent DNA cleavage by restriction endonuclease IIS [6]. Gopal and Bhagwat [7] suggested a method of identification of methylated base based on the use of E. coli exonuclease III. Along with radioactive SAM, this method suggests synthesis of DNA duplexes containing the phosphothioate internucleotide group. Finally, the methylated nucleoside can be identified using mass-spectrometry. This method requires special equipment [8].
We have developed an alternative biochemical method for determination of a methylated deoxycytidine position based on the bisulfite reaction; the latter can be used to identify non-methylated dC residues from several residues of the recognition site. The bisulfite reaction together with polymerase chain reaction is often used for evaluation of the methylation status of CpG islands in DNA and for mapping of all methylated dC residues in a known nucleotide sequence [9, 10]. Sodium bisulfite causes deamination of dC residues in single-stranded DNA with formation of deoxyuridine (dU) residues, leaving 5-methyldeoxycytidine and N4-methyldeoxycytidine residues unchanged [11]. The dU residues can be easily detected with the repair enzyme uracil-DNA glycosylase (UDG). Short DNA substrates can be used in our method, which is especially suitable for methyltransferases recognizing a nucleotide sequence containing only two dC. We have successfully applied this method for study of DNA-methyltransferase NlaX (M.NlaX) from Neiserria lactamica [12]. This enzyme does not have a tandem restriction endonuclease, and neither the nucleotide sequence of the methylation site nor the position of the methylated dC residue was known for it.
MATERIALS AND METHODS
Oligodeoxyribonucleotides used in this work were synthesized by the standard amidophosphite method in an automatic synthesizer (Applied Biosystems 380B, USA). Oligodeoxyribonucleotides containing 5-fluoro-2´-deoxycytidine were obtained as described earlier [13].
Recombinant forms of M.SsoII, M.NlaX, and R.SsoII having six histidine residues at the N-end were isolated via a two-step method using chromatography on heparin-Sepharose and Ni-NTA-agarose [14]. Uracil-DNA glycosylase from E. coli and T4 polynucleotide kinase are commercial preparations from New England BioLabs (USA) and Sibenzyme (Russia), respectively.
The following reagents were used in this study: EDTA, (NH4)2SO4, LiClO4, and formamide from Fluka (Switzerland); N,N´-methylenebisacrylamide, N,N,N´,N´-tetramethylethylenediamine, acrylamide, and glycerol from Serva (Germany); SDS and dye markers Bromophenol Blue (BPB) and Xylene Cyanole (XC) from Reanal (Hungary); dithiothreitol, Tris-HCl, piperidine, formic acid, dimethylsulfate, hydrazine, potassium permanganate and sodium metabisulfite from Merck (Germany); hydroquinone from Sigma (USA); [gamma-32P]ATP with specific radioactivity 1000 Ci/mole from Izotop (Russia). Other reagents used were extra pure grade products produced in Russia.
Radioactive label was inserted into oligonucleotides by T4 polynucleotide kinase phage in the presence of [gamma-32P]ATP. The 32P-labeled products were separated by electrophoresis in 20% polyacrylamide gel in Tris-borate buffer (50 mM Tris-HCl, 50 mM H3BO3, 1 mM EDTA, pH 8.3) in the presence of 7 M urea and then detected by autoradiography. 5´-[32P]oligonucleotides were isolated from the gel by 2 M LiClO4 for 18 h at room temperature with subsequent precipitation by acetone.
Solutions of DNA duplexes were prepared by mixing of the appropriate oligonucleotides in equimolar ratios. 5´-Phosphorylated duplexes with known specific radioactivity were obtained by addition of the appropriate 5´-[32P]oligonucleotide to a specific amount of DNA duplex and subsequent annealing.
The efficiency of interaction between methyltransferases NlaX and SsoII (M.SsoII) and DNA duplexes was evaluated via the degree of the substrate protection from hydrolysis by restriction endonuclease SsoII (R.SsoII). DNA duplex (3.5 pM) with one 32P-labeled strand was incubated with M.NlaX or M.SsoII (10 activity units) in 50 mM Tris-HCl buffer, pH 7.6, containing 10 mM EDTA, 7 mM 2-mercaptoethanol, and 0.1 mM SAM for 2 h at 37°C. After the reaction with methyltransferases, oligonucleotides were precipitated by acetone, dried, annealed in 50 mM Tris-HCl buffer, pH 7.6, containing 50 mM NaCl, 10 mM EDTA, and 7 mM 2-mercaptoethanol and incubated with R.SsoII (10 activity units) for 1 h at 37°C. The products of hydrolysis were analyzed by electrophoresis in 20% polyacrylamide gel containing 7 M urea.
The non-methylated deoxycytidine residue in [32P]oligonucleotide 5´-AGAGCCAGGTTGGC-3´ incorporated into the duplex III was determined according to a specially developed method: 100 µl of 5 M sodium bisulfite (2.5 M sodium metabisulfite and 125 mM hydroquinone, pH 5.0) were added to the preliminarily precipitated oligonucleotide (~10-100 nM in solution) and incubated for 4 h at 50°C in the dark. Then the reaction mixture was desalted by gel filtration on a NAP-5 column (Amersham-Pharmacia-Biotech, Sweden) and treated with 0.3 M alkali for 15 min at 37°C (volume 60 µl). The solution was neutralized by addition of 3 M ammonium acetate to pH 7.0. Oligonucleotides were precipitated with ethanol, dried, and dissolved in 20 mM Tris-HCl buffer, pH 7.5, containing 60 mM NaCl, 10 mM EDTA and 0.5 mg/ml BSA. UDG (2 activity units) was added to the reaction mixture, and the latter was incubated for 1 h at 37°C. The samples were dried and treated with 10% aqueous solution of piperidine for 30 min at 90°C. After removal of traces of piperidine by re-evaporation with water, the DNA fragments were separated by electrophoresis in 20% polyacrylamide gel containing 7 M urea.
Cross-linking of DNA methyltransferases to DNA duplexes containing 5-fluoro-2´-deoxycytidine was performed in 50 mM Tris-HCl buffer, pH 7.6, containing 10 mM EDTA, 7 mM 2-mercaptoethanol, and 0.1 mM SAM. The 32P-labeled in the modified strand DNA duplex IV or V (2 pM) was incubated with 70 pM M.NlaX or M.SsoII (~4 µg) for 1 h at 37°C. After heating in denaturing solution (2% SDS, 0.05% mercaptoethanol, 7 M urea), the reaction products were analyzed by electrophoresis in 12% polyacrylamide gel containing 0.1% SDS according to Laemmli [15] and visualized by autoradiography.
RESULTS AND DISCUSSION
Determination of the M.NlaX recognition site. Our first goal was to determine the nucleotide sequence of the M.NlaX recognition site. Preliminary investigations demonstrated that the amino acid sequence of M.NlaX is highly homological with the two methyltransferases SsoII and ScrFI recognizing a pentanucleotide sequence 5´.CCNGG.3´ (where N = C, G, A, or T) in the double-stranded DNA [14]. This fact suggested that M.NlaX recognizes a similar and maybe analogous site. To test this hypothesis, we used two 14-membered DNA duplexes; their sequence included a 5´.CCNGG.3´ fragment, and DNA duplex I had dA/dT residues as a central nucleotide pair and DNA duplex II had dG/dC residues:
5´ AGAGCCAGGTTGGC 3´ 3´ TCTCGGTCCAACCG 5´ (I) 5´ AGAGCCGGGTTGGC 3´ 3´ TCTCGGCCCAACCG 5´ (II)
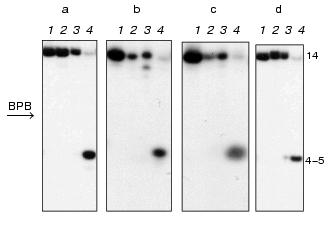
DNA duplexes I and II 32P-labeled at the 5´-end of the upper or lower strand were incubated with M.NlaX in the presence of SAM cofactor. The reaction mixture was treated by restriction endonuclease SsoII in the presence of Mg2+. This enzyme cleaves the double-stranded sequence 5´.vCCNGG.3´ at the position shown by the arrow. R.SsoII is unable to hydrolyze the recognition site if the internal or external dC residue is methylated [16, 17]. In the control, duplexes I and II were modified by M.SsoII and incubated with R.SsoII. Autoradiograms of electrophoretic separation of the products of hydrolysis of the upper and lower DNA duplexes I (a, b) and II (c, d), respectively, are presented in Fig. 1. As shown, the interaction of both methyltransferases with duplexes I and II prevents hydrolysis of the latter by R.SsoII. Consequently, similar to M.SsoII, M.NlaX recognizes 5´.CCA/TGG.3´ and 5´.CCG/CGG.3´ nucleotide sequences and methylates their internal or external cytosine.
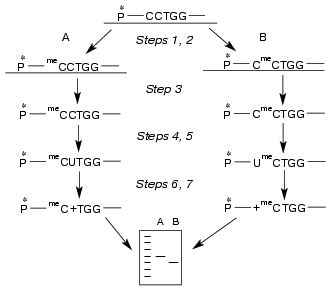
Determination of non-methylated dC residue in M.NlaX recognition site. Which of two cytosines of CCNGG sequence is methylated by M.NlaX? To answer this question, it is sufficient to detect a non-methylated base. In order to determine the position of the non-methylated cytosine in M.NlaX, we developed an original method including sequential use of the bisulfite reaction and repair enzyme uracil-DNA glycosylase. The scheme of this method is shown in Fig. 2. The DNA duplex having a methyltransferase recognition site is incubated with the studied enzyme in the presence of SAM to complete methylation of the substrate (step 1). To facilitate separation of the strands in subsequent steps of the experiment, one of the strands of the substrate should be several nucleotide units longer than the other. For removal of non-methylated molecules, the reaction mixture is then treated by RSsoII (step 2). The strands of DNA duplex are separated by gel electrophoresis under denaturing conditions (step 3). The methylated oligonucleotides are isolated from the gel, treated by sodium bisulfite (step 4), desalted, and incubated with alkali (step 5). Sodium bisulfite is reversibly added to the C5-C6 double bond of cytosine, forming 5,6-dihydro-6-sulfonate, which is rapidly deaminated on alkali treatment with subsequent elimination of bisulfite [18, 19]. The dU-containing oligonucleotide thus obtained is incubated with UDG; this results in excision of uracil with formation of the apyrimidine site (step 6). After hydrolysis by 10% aqueous piperidine solution at the place of modification, the reaction mixture is separated by gel electrophoresis under the denaturing conditions (step 7). To determine the length of the formed oligonucleotide, it is convenient to use as markers the fragments of the initial oligonucleotide obtained according to the modified Maxam-Gilbert procedure [20]. The only limitation on our approach is the need for design of the substrates in which the dC residues are absent from the flanking sequence adjacent to the 5´-end of the recognition site of the methyltransferase.Fig. 1. Electrophoretic analysis of the products of hydrolysis of DNA duplexes I ([32P] label in the upper (a) and in the lower (b) strand) and II ([32P] label in the upper (c) and in the lower (d) strand) after treatment with R.SsoII. Lanes: 1) initial duplex (control); 2, 3) hydrolysis of the duplexes methylated by M.NlaX and M.SsoII, respectively; 4) hydrolysis of non-methylated duplex. Position of the marker Bromophenol Blue (BPB) is shown by the arrow. On the right, the length of oligonucleotide fragments.
To determine position of the non-methylated M.NlaX cytidine residue, we used a 14/18-membered DNA duplex (III) containing the CCA/TGG nucleotide sequence:Fig. 2. Scheme of the method for determination of non-methylated dC residue in M.NlaX recognition site including sequential use of bisulfite reaction and UDG. Two possible ways of methylation (A, B) are considered. All the steps are described in the text. P* is 32P.
5´ AGAGCCAGGTTGGC 3´ 3´ AGTCTCGGTCCAACCGAG 5´ (III)
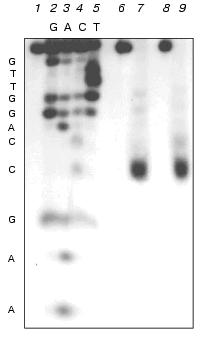
Analogous experiments were also performed with M.SsoII; the latter was found to methylate the internal dC residue of the recognition site, forming 5-methylcytosine [16]. As shown in Fig. 3, in experiments with M.NlaX as well as with M.SsoII, the fragments of the same length are formed; they correspond to the splitting of the 14-membered oligonucleotide from duplex III at the external dC residue of the recognition site, that is, the latter is not methylated by M.NlaX. The internal cytosine is obviously to be modified.
Identification of the type of methylation of internal dC residue of M.NlaX. To prove our data on the position of the modified cytosine and to identify the place of methylation in it, we used DNA duplexes IV and V containing 5-fluoro-2´-deoxycytidine (fl5dC, F) instead of the internal dC residue [21]:Fig. 3. Determination of the length of oligonucleotide fragment after sequential treatment by sodium bisulfite, UDG and 10% piperidine solution of 14-membered strand of DNA duplex III pre-methylated with M.NlaX (lane 7) and M.SsoII (lane 9). Lanes: 2-5) separation of the products of chemical modification according to Maxam-Gilbert of non-methylated 14-membered strand of duplex III; 1) initial non-methylated 14-membered oligonucleotide after treatment by 10% piperidine; 6-8) after treatment by sodium bisulfite, UDG, and 10% piperidine sequentially.
5´ AGAGCFAGGTTGGC 3´ 3´ AGTCTCGGTCCAACCGAG 5´ (IV) 5´ AGAGCCAGGTTGGC 3´ 3´ AGTCTCGGTFCAACCGAG 5´ (V)
On modification of the fifth position of the dC residue by methyltransferase, an intermediate covalent complex between enzyme and substrate is formed. Change of the methylated dC residue by fl5dC makes this formation irreversible [21] and easily detectable by gel electrophoresis according to Laemmli.
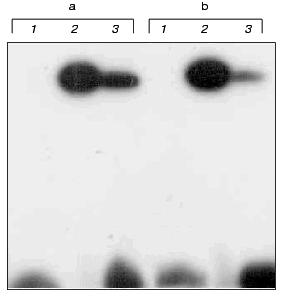
The DNA duplexes IV and V 32P-labeled at the 5´-end of the strand containing the fl5dC residue were incubated with M.NlaX (and M.SsoII as a control) in the presence of SAM. Then the reaction mixtures were analyzed by SDS-PAGE. For both M.NlaX and M.SsoII, a covalent complex was formed with each of fl5dC-containing oligodeoxyribonucleotides (Fig. 4). The results indicate that in the CCNGG pentanucleotide sequence M.NlaX methylates the internal dC residue at the fifth position of the heterocycle.
Thus, we determined the specificity of M.NlaX. This is the first time the bisulfite reaction in combination with the repair enzyme uracil-DNA glycosylase has been suggested for identification of a non-methylated dC residue.Fig. 4. Electrophoretic analysis of cross-linking of M.SsoII (lanes 2) and M.NlaX (lanes 3) to DNA duplexes IV (a) and V (b). Lanes 1: initial DNA duplexes IV and V.
The authors are grateful to Dr. E. A. Romanova (Moscow State University) for the synthesis of oligodeoxyribonucleotides and to Dr. A. E. Sud'ina and M. B. Viryasov (Moscow State University) for help in preparation of the figures. This work was supported by grants of the programs “Russian Universities” (No. 015.07.02.027), “Leading Scientific Schools” (No. 00-15-97944), and of the Russian Foundation for Basic Research (No. 01-04-48605).
REFERENCES
1.Li, E., Beard, C., and Jaenisch, R. (1993)
Nature, 366, 362-364.
2.Razin, A., and Shemer, R. (1995) Hum. Mol.
Genet., 4, 1751-1755.
3.Noyer-Weidner, M., and Trautner, T. A. (1993) in
DNA Methylation: Molecular Biology and Biological Significance
(Jost, J. P., and Saluz, H. P., eds.) Birkhauster Verlag, Basel, pp.
39-108.
4.Boyer, H. W., Chow, L. T., Dugaczyk, A., Hedgepeth,
J., and Goodman, H. M. (1973) Nature New Biol., 224,
40-43.
5.May, M. S., and Hattman, S. (1975) J.
Bacteriol., 122, 129-138.
6.Posfai, G., and Szybalski, W. (1988) Gene,
69, 147-151.
7.Gopal, J., and Bhagwat, A. S. (1995) Nucleic
Acids Res., 23, 29-35.
8.Tamura, T., Araki, Y., Yamaoka, S., Inagaki, K.,
and Tanaka, H. (1997) Nucleic Acids Res., 25,
4162-4164.
9.Herman, J. G., Graff, J. R., Myohanen, S., Nelkin,
B. D., and Baylin, S. B. (1996) Proc. Natl. Acad. Sci. USA,
93, 9821-9826.
10.Clark, C. J., Harrison, J., Paul, C. L., and
Frommer, M. (1994) Nucleic Acids Res., 22, 2290-2297.
11.Vilkaitis, G., and Klimasauskas, S. (1999)
Analyt. Biochem., 271, 116-119.
12.Lau, P. C. K., Forghani, F., Labbe, D., Bergeron,
H., Brousseau, R., and Holtke, H. J. (1994) Mol. Gen. Genet.,
243, 24-31.
13.Wyszynski, M. W., Gabbara, S., Kubareva, E. A.,
Romanova, E. A., Oretskaya, T. S., Gromova, E. S., Shabarova, Z. A.,
and Bhagwat, A. S. (1993) Nucleic Acids Res., 21,
295-301.
14.Karyagina, A. S., Lunin, V. G., Levtchenko, I.
Ya., Labbe, D., Brousseau, R., Lau, P. C. K., and Nikolskaya, I. I.
(1995) Gene, 157, 93-96.
15.Laemmli, U. K. (1970) Nature, 227,
680-685.
16.Nikol'skaya, I. I., Lopatina, N. G., Sushkov, S.
V., and Debov, S. S. (1985) Biokhimiya, 50,
1694-1700.
17.Gromova, E. A., Kubareva, E. A., Elov, A. A.,
Akatova, E. A., Nikol'skaya, I. I., and Shabarova, Z. A. (1991)
Biokhimiya, 56, 552-559.
18.Shapiro, R., DiFate, V., and Welcher, M. (1974)
J. Am. Chem. Soc., 96, 906-912.
19.Wang, R. Y.-H., Gehrke, C. W., and Ehrlich, M.
(1980) Nucleic Acids Res., 8, 4777-4790.
20.Maxam, A., and Gilbert, W. (1977) Proc. Natl.
Acad. Sci. USA, 74, 560-564.
21.Santi, D. V., Garrett, C. E., and Barr, P. J.
(1983) Cell, 33, 9-10.