Respiratory Complex I: Structure, Redox Components, and Possible Mechanisms of Energy Transduction
A. D. Vinogradov
Department of Biochemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3955; E-mail: adv@biochem.bio.msu.su
Received May 18, 2001
Structural arrangements and properties of redox components of the mitochondrial and bacterial proton-translocating NADH:quinone oxidoreductases are briefly described. A model for the mechanism of proton translocation at first coupling site, which emphasizes participation of specifically Complex I-associated ubisemiquinones, is discussed. An alternative mechanism is proposed where all redox reactions take place in a hydrophilic part of the enzyme and the free energy accumulated as conformational constraint drives the proton pump associated with the hydrophobic polypeptides.
KEY WORDS: NADH:quinone oxidoreductase, Complex I, respiratory chain, proton translocation, electrochemical coupling
INTRODUCTION
The metabolic breakdown of proteins, carbohydrates, and lipids, which provides cells with energy, requires continuous regeneration of oxidized NAD+, the major collector of reducing equivalents. In mammalian cells and in most microorganisms oxidation of NADH is catalyzed by the respiratory chains (NADH oxidases), the oligoenzymatic complex localized in the inner mitochondrial membranes or in the bacterial plasma membranes. The respiratory chains regenerate NAD+ by oxidizing NADH by a terminal electron acceptor (oxygen in aerobic organisms) to yield free energy that is accumulated as a gradient of electrochemical potentials of hydrogen ions (Delta µH+) or sodium ions (Delta µNa+ in some prokaryotes) across the coupling membrane impermeable for ions. In the late sixties of the 20th century, it was established that synthesis of ATP coupled with the electron transfer occurs at three distinct sites of the respiratory chain: NADH:quinone oxidoreductase (Site 1), ubiquinol:cytochrome c oxidoreductase (Site 2), and cytochrome c oxidase (Site 3). By this time, D. Green and his associates had succeeded in resolving the respiratory chain into individual lipoprotein complexes catalyzing the redox reactions corresponding to all three coupling sites. These “enzymes” were named Complexes I, III, and IV, respectively. When individual complexes are mixed under certain conditions (concentrated solutions, removal of the detergents used for solubilization, addition of cytochrome c), NADH oxidase activity is restored.
In the late seventies, most scholars in the field accepted the mechanism of energy transformation in the respiratory chain in terms of Mitchell's electrochemical coupling theory [1, 2].
Recently, complete atomic space structure of cytochrome c oxidase (Complex IV) and ubiquinol:cytochrome c oxidoreductase (Complex III) have been established and experiments aimed to verify any working hypotheses on the electrochemical coupling mechanism catalyzed by these complexes reached a new stage. As far as Complex I is concerned (NADH:ubiquinol oxidoreductase, Type 1 NADH dehydrogenase of prokaryote, coupling Site 1, all these terms refer to the same genetically and functionally related enzymes), it is hard to deny that very little is known about the structure, intramolecular electron transfer sequence between potentially active redox components, and the mechanism of electrochemical coupling.
By the late eighties interest in studies on Complex I were considerably revived. Current knowledge on the enzyme structure [3, 4], its molecular evolution [5, 6], catalytic and regulatory properties [7], redox components [8, 9], and its possible involvement in the development of pathologies [10, 11] have been summarized in a number of recent reviews. Much less information concerning the molecular mechanism of energy transduction is available. This is because only a few studies aimed directly at the energy transduction mechanism have appeared in the current literature. In this short review the data that in my opinion are most significant for possible mechanisms of energy transduction will be briefly summarized.
THE ENZYME STRUCTURE
Bovine heart mitochondrial Complex I is a giant (on the enzymology scale) construction composed of at least 43 different polypeptides with a total molecular mass of about 1 million daltons [4]. Seven polypeptides are encoded by the mitochondrial genome [12], and their homologs are found in the much simpler prokaryotic Type 1 NADH dehydrogenases (13-14 different polypeptides) [13, 14] and also in fungal Neurospora crassa Complex I (not less than 35 identified different subunits) [15]. The reconstruction of electron microscopy images of isolated N. crassa enzyme shows that Complex I looks like a “boot” immersed into the phospholipid membrane in such a way that the “bootleg” forms a peripheral part exposed to the mitochondrial matrix [16]. This boot-like structure is evolutionarily conserved; preparations of E. coli Type 1 NADH dehydrogenase (13 polypeptides) appear as a “boot” of smaller size [17]. The amino-acid sequences of the mitochondrially-encoded polypeptides suggest the presence of 3-14 hydrophobic alpha-helixes in each of them (up to 53 in total) [4]. Several nuclearly encoded subunits also contain hydrophobic alpha-helixes [18]. In mitochondrial or bacterial membranes (inside-out particles), Complex I catalyzes the NADH oxidase or NADH:quinone-acceptor reductase reactions which are sensitive to the specific inhibitors rotenone and piericidine A [19-21] and to a number of other hydrophobic chemically simple (Triton X-100 [22]) or very complex compounds [23, 24]. Most of the preparations, obtained by either solubilization of the membranes or by further resolution of the classical rotenone-sensitive Hatefi's Complex I [25], catalyze rotenone-insensitive NADH oxidation by a number of artificial electron acceptors [26]. It is generally believed that all rotenone-insensitive NADH dehydrogenase preparations are fragments of Complex I.
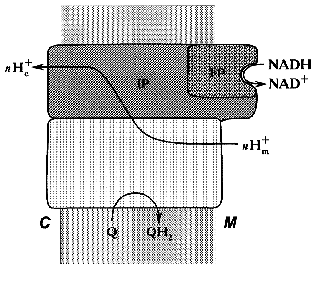
Treatment of the bovine heart mitochondrial Complex I (the preparation is “soluble” only in the presence of detergents) with chaotropic agents such as sodium perchlorate results in solubilization of about 1/3 of the protein. The soluble fraction thus obtained catalyzes rotenone-insensitive NADH oxidation, and further fractionation results in purification of a “minimal” catalytically active fragment, so-called FP (flavoprotein) containing flavin and non-heme iron and composed of only three subunits (51, 24, and 10 kD) [27]. The other part of the perchlorate-solubilized fraction, so-called IP (iron-protein) is enriched in iron and sulfur content. It is composed of at least six polypeptides and shows no catalytic activity. The fragment that remains water-insoluble after extraction of the original material by sodium perchlorate is called HP (hydrophobic protein) and it also has no any catalytic activity. Treatment of submitochondrial particles with phospholipase results in solubilization of so-called high molecular mass rotenone-insensitive NADH dehydrogenase [28], which is similar to a combination of FP + IP. The fractionation of bovine heart mitochondrial Complex I in the presence of detergents leads to separation of two fragments: Ialpha (catalytically active) and Ibeta (catalytically inert) [29]. Treatment of N. crassa Complex I with sodium bromide results in separation of the catalytically active fragment (“bootleg”, FP + IP) and the hydrophobic part (HP) [30]. The generalized structure of Complex I within the coupling membrane is shown schematically in Fig. 1.
Fig. 1. Schematic presentation of the structure of Complex I within the inner mitochondrial membrane. M, matrix; C, intermembranous space; FP, IP, and HP, the subfragments of bovine heart enzyme separated after Complex I fractionation.
REDOX COMPONENTS
Multiple redox components (FMN, several iron-sulfur clusters, and tightly bound ubiquinone) potentially capable of intramolecular electron transfer from the enzyme-bound NADH to the terminal quinone acceptor are found in Complex I. Before discussing properties of the components, some general comments seem reasonable to make. Optical absorption spectra of flavins and iron-sulfur clusters overlap and light spectroscopy is difficult to use for studies of electron transfer reactions even in purified Complex I preparations. The only available method for detection of redox transitions in the iron-sulfur clusters and also in the reactions coupled with a formation of the free radical forms of flavo- and ubisemiquinones is EPR spectroscopy. This unique technique, when applied for standard biochemical studies, is somewhat limited. First, no parameter equivalent to the molar absorption coefficient used for quantifications in optical spectroscopy exists in EPR-spectroscopy. The observed signal amplitudes depend on a number of factors (temperature, microwave power, etc.) and only approximate quantification of unpaired electron concentration (content) can be done in most cases. Second, the signal/noise ratio in modern EPR spectrometers allows reliable detection of about 10-6 “moles per liter” of unpaired electrons (without accumulation of the signal by multiple repeated scans). This results in at least two further difficulties. The enzyme concentration (protein-lipid-detergent complex or suspension of particles) in EPR-samples is usually several orders of magnitude higher than that conventionally used for enzymatic activity assays. Thus, for example, isolated Complex I in EPR samples is present under conditions where its catalytic activity is either strongly modified or almost completely inhibited by a detergent. Another complication is difficulty in registration of the redox reactions under steady-state conditions. The turnover number of Complex I in uncoupled submitochondrial or sub-bacterial particles at 25°C is about 104 min-1 [7]. At protein content in the samples of about 20 mg/ml (~10-6 M) and at reasonable concentration of the substrates (NADH, oxygen, exogenously added quinone) they are converted to the products within several seconds after initiation of the reaction. The registration of EPR-detectable components under steady-state conditions is difficult when simple conventional mixing of the samples is used.
The last comment to be made is that EPR spectrometers are expensive and their use and meaningful interpretation of the data particularly require rather special knowledge. An obvious consequence is that cryogenic EPR technique is a “monopolized” area, and the data obtained in one laboratory are not easy to confirm or disapprove in another.
This preliminary notes are intended to raise great precautions when the results obtained by low temperature EPR spectroscopy are to be analyzed. This is certainly true for what is described in this mini-review.
FLAVIN
All preparations of Complex I and the catalytically active fragments derived from them contain flavin mononucleotide (FMN). Presumably 1 mol of enzyme contains 1 mol of flavin. It should be clearly understood, however, what the definition of “one mole of enzyme” is. The possibility that Complex I operates as homodi- or homooligomer within the mitochondrial membranes cannot be excluded. Variously arranged heterodimeric structures of Complex I have been discussed by Albracht and his associates [31-33], although in my opinion none of these models can account for all the data available. The highly purified rotenone- or piericidine-sensitive preparations of Complex I contain 1.2-1.5 nmol of FMN per mg protein [34]. The theoretically calculated molecular mass of bovine heart enzyme (taken as a sum of the individual subunits molecular masses assuming their 1 : 1 ratio) is 880 kD [4]. Thus, the expected content of FMN in homogeneous protein is of about 1.2 nmol per mg protein, a value closely corresponding to that determined analytically. Clearly, the same content of FMN is expected for any homooligomer. FMN content in highly purified three-subunit fragment FP (~10 nmol per mg protein) is also consistent with that theoretically expected [35]. It should be noted that 51 kD subunit, the most conceivable candidate for FMN binding, is present in 1 : 1 stoichiometry with other subunits of FP + IP fraction [36]. Thus, it appears that 1 mol of monomeric Complex I contains 1 mol of FMN.
FMN is tightly but noncovalently bound to the protein and no loss of flavin occurs under rather drastic treatments such as exposure to chaotropic agents, detergents, and solubilization by acidic ethanol. The reduction of FP in very diluted solution (~10-9 M) results in loss of the catalytic activity, which is prevented in concentrated enzyme solutions or by the addition of FMN [37]. The degree of inactivation (FMN dissociation) depends on flavin reduction as expected if the midpoint redox potential for the FMNox <--> FMNred transition at pH 8.0, 25°C would be -320 mV [37], a value which is substantially lower than that for free FMN (approximately -280 mV [38, 39]). In other words, oxidized FMN binds to the protein several orders of magnitude tighter than does reduced FMN. This finding suggests a reorganization of the chemical bonds that keep flavin in the protein; thus, significant conformational change of the protein is expected upon oxidoreduction of FMN. Indeed, alterations of the intersubunit contacts upon reduction of the enzyme have been reported [40]. It worth noting that FP is only a small fragment of Complex I, and its catalytic capacity and possibly properties of FMN are significantly different from those in intact Complex I [41]. The thermodynamic properties of FMN in isolated Complex I have been studied by low-temperature EPR as flavosemiquinone appearance upon redox titration by artificial dyes or by the substrate nucleotides [42]. The midpoint redox potential of -400 mV (pH 8.2, 25°C) for two-electron reduction of FMN by artificial electron donors was found. The potential was shifted to ~-300 mV when FMN radical was titrated by the nucleotide substrates. The flavosemiquinone free radical observed in the presence of artificial reductants was somewhat different from that which is seen for free FMN or FMN in flavodoxin [43]. The EPR-signal was slightly broader and its amplitude was not saturated at high microwave power, thus suggesting an interaction of FMN free radical with some closely located rapidly relaxing paramagnetic center. Free radical seen in the presence of NADH/NAD+ (at higher redox potential) was significantly narrower, indicating different interaction between flavosemiquinone and its paramagnetic “neighbor”. FMN is most likely bound to 51 kD subunit: the substitution of 51 kD subunit in N. crassa enzyme for a mutated defective copy results in assembling of complex identical to the wild type, but completely devoid of flavin, one iron-sulfur cluster (N-3), and the NADH binding and oxidizing capacities [44].
The following conclusions can be made to summarize the data on FMN in Complex I. The nucleotide is bound in the hydrophilic, matrix-exposed part of the enzyme and serves as the primary electron acceptor for NADH. Its midpoint redox potential suggests that no “coupling site” is possible between NADH and FMN. The oxidoreduction of FMN during catalysis is likely to be accompanied by structural rearrangement of the enzyme subunits, at least in its hydrophilic (FP + IP) part.
IRON-SULFUR CENTERS
Various preparations of bovine heart mitochondrial Complex I contain 11-28 atoms of non-heme iron per mol FMN [34] and about the same amount of sulfur easily liberated as hydrogen sulfide at acid pH (acid-labile sulfur). The results of chemical analysis thus suggest the presence of several iron-sulfur clusters in the enzyme. Detailed studies on the preparations at different degrees of resolution have revealed the presence of at least six paramagnetic iron-sulfur centers different in their redox potentials and EPR characteristics (signal shape, saturation by microwave power, temperature dependencies). The characteristics of iron-sulfur centers in Complexes I of mammalian mitochondria, prokaryotes, and N. crassa have been described by the experts in low-temperature EPR [8, 9, 45]. Here I will describe them only briefly. It should be emphasized that the midpoint redox potentials of the iron-sulfur centers (see table), the parameter crucially important for possible sequence of electron transfer, depend on purification (degradation?) of particular preparation and also on the titration procedure (NAD+/NADH-, succinate/fumarate-couple, the presence of redox mediators).
Redox components of Complex I
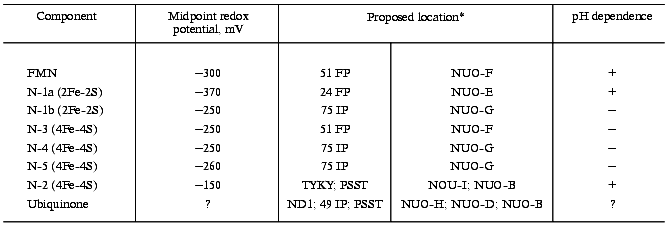
*Left column, subunits indicated according to the
nomenclature accepted for bovine heart enzyme [4,
13, 14, 18]. This nomenclature is rather arbitrary, some
subunits being named according to their fractionation (FP, IP
fractions) and apparent molecular mass; the subunits that are encoded
by the mitochondrial genome are designated as ND; N-terminal unmodified
subunits are designated by four N-terminal amino acid residues (one
letter code); the nuclear encoded N-terminal modified subunits are
designated by B and their molecular mass (for example B15). Right
column, homologs subunits of prokaryotic Type 1 NADH dehydrogenase. For
prokaryotic enzyme the subunits are designated as NUO and by a letter
in alphabetical order corresponding to the position of particular gene
in the nuo operon (n, NADH, u, ubiquinone, and
o, oxidoreductase).
The enzyme bears two binuclear and four tetranuclear iron-sulfur centers (table). The total amount of iron per mol of the enzyme (mol FMN) calculated as the sum of iron content in two binuclear and four tetranuclear iron-sulfur centers is equal to 20, whereas chemical analysis of the most purified preparations reveals up to 28 atoms of iron per mol of FMN. Thus, it cannot be excluded that some other EPR-undetectable centers exist in addition to those indicated in the table. The redox potential of one binuclear center (N-1a) is so negative that it can not be reduced by NADH. The potentials of two centers (N-1a and N-2) are pH dependent (-60 mV per pH unit at pH 6.2-8.7) [46] indicating that their one-electron oxidoreduction is coupled to protonation/deprotonation. This does not mean that the redox reaction is accompanied by protonation/deprotonation of (Fe-S) cluster per se, it suggests rather that protonation of some group which can be located close to or far away from the cluster is obligately coupled with oxidoreduction. This note is of importance for discussion of the mechanism of vectorial proton translocation.
Four of six iron-sulfur centers (N-1b, N-3, N-4, N-5) form an almost isopotential group and the only significant redox gap exists between these four clusters and most positive N-2. This suggests that N-2 is the most possible candidate for the terminal electron transfer to bulk ubiquinone (redox potential of the Q/QH2 couple is about +100 mV [47]). Since the redox potential of N-2 is pH dependent, this iron-sulfur center is usually considered as an immediate participant of the coupling mechanisms in a number of hypothetical schemes [48]. It is worth noting that the redox potential of N-2 at pH 7.0 was reported either as -20 mV (ultrasonic particles from pigeon heart mitochondria) or as -140 mV or -20 mV (bovine heart mitochondria) in the earlier original studies [46, 49]. Nevertheless, in most reviews published before the early nineties only the value of -20 mV was taken as the midpoint redox potential of N-2. Consequently, the possibility of the energy-coupling mechanism operating between N-2 and ubiquinone has not been discussed for thermodynamic reasons. We have reported that in uncoupled bovine heart submitochondrial particles N-2 is not reduced at potentials higher than -120 mV (succinate/fumarate couple) and this was the key point for postulating the proton translocating mechanism coupled with electron transfer between N-2 and ubiquinone [50-52] (see section “Electrochemical Coupling”).
The set of iron-sulfur clusters in Complex I is evolutionarily conserved. Iron-sulfur centers with similar characteristics are found in the membranes of P. denitrificans, Rb. sphaeroides, T. thermophilus, and E. coli [45]. It should be remembered that Type-1 NADH dehydrogenases, which are equivalent to the mitochondrial Complex I, are composed of only 14 subunits. This equivalency should not, however, be considered too literally. For example, binuclear, pH-dependent center N-1a in P. denitrificans is [53] significantly more positive (-150 mV, pH 7.0) than its counterpart in mitochondria (-370 mV, pH 7.0) [46]. E. coli membranes contain additional center N-1c with redox potential of -240 mV [54]. In T. thermophilus membranes, the potential of the tetranuclear N-2 center that is slightly different in its line shape is significantly more negative (-300 mV) and does not depend on pH [45]. Interestingly, the membranes of this organism contain menaquinone (Em,7 = -75 mV) instead of ubiquinone (Em,7 = +100 mV).
Under steady-state NADH oxidation all iron-sulfur clusters are almost completely reduced [51, 55] and no clear “cross-over” point is seen between the clusters upon the transition from tightly coupled to completely uncoupled states [51].
The identification of genes for all the subunits of bacterial proton-translocating NADH dehydrogenases, EPR studies of their iron-sulfur centers, and sequencing of all mitochondrial Complex I subunits have provided extremely powerful tools for localization of Fe-S centers within the enzyme structure. If a subunit of Complex I contain “motifs” of cysteine residues typical for binuclear or tetranuclear clusters in their amino-acid sequences (cysteine is the most common ligand for iron in iron-sulfur clusters) and this subunit (and motif) is evolutionary conserved, it can be safely concluded that this subunit bears the iron-sulfur center. This approach has reached the state where several individual subunits were overexpressed, purified, and their iron-sulfur cluster(s) were chemically reconstituted (incubation of protein in the presence of Fe2+, sulfide, and mercaptoethanol). The other approach to locate Fe-S clusters is to resolve Complex I into its fragments and studies on their chemical composition and EPR properties. All these studies make it very likely that all the clusters are located in a hydrophilic part of the enzyme.
BOUND UBIQUINONE
Rotenone-sensitive Complex I prepared by the classical Hatefi's procedure contains about 4 mol of ubiquinone per mol FMN [25, 56]. This finding as such provides little information because ubiquinone is very hydrophobic and any hydrophobic compound which is present in great excess over any other component of the respiratory chain is expected to be bound to the protein-detergent-phospholipid complex (respiratory complexes I-IV). However, if this binding is specific, the protein environment may stabilize intermediate semiquinone forms. An intermediate formation of free radicals of ubisemiquinone during electron transfer within Complex I is almost obligatory because all iron-sulfur clusters are one-electron reductants. The equilibrium of ubisemiquinone dismutation reactions in solution is greatly shifted to the formation of its fully oxidized and reduced forms, thus detection of ubisemiquinone quantitatively comparable with the amount of potentially capable binding sites strongly suggests specific and functionally important binding.
To my knowledge, Suzuki and King were the first to report the presence of ubisemiquinone free radical in purified Complex I [56]. They observed a narrow g = 2.00 EPR-signal arising after the addition of NADH to the “solution” of Complex I (~30 mg per ml) at room temperature. The signal amplitude was dependent on the concentration of NADH and rotenone [56].
Later, using different preparations (tightly coupled submitochondrial particles [57]) and different approach (rapid freezing of the samples catalyzing steady-state NADH oxidation followed by analysis of the EPR signals at low temperature) the specifically bound Complex I associated ubisemiquinone was discovered in our laboratory [50, 51]. First attempts to reproduce our results in other laboratory have failed [58], evidently because uncoupled and de-activated submitochondrial particles were used (see [26] for description of activation/de-activation phenomenon in Complex I). Later, the presence of Complex I-associated ubisemiquinone in such submitochondrial particles was confirmed in the same laboratory [59], and in a series of studies by Albracht's group [59-61] and by our group in collaboration with Ohnishi's laboratory [62-64] substantial information about a “new” redox component of the enzyme have accumulated. The properties of Complex I-associated ubisemiquinones are briefly summarized below.
The semiquinone signal in the particles oxidizing succinate seen at low temperature (~40 K) disappears in the presence of rotenone (inhibitor of Delta µH+-dependent reverse electron transfer) or after Complex I de-activation [5, 64]. The microwave power saturation behavior suggests an interaction between the ubisemiquinone free radical and a rapidly relaxing paramagnetic center (iron-sulfur cluster N-2) [50, 63]. More detailed analysis of the power-saturation curves reveals the presence of at least two forms (states) of Complex I-associated ubisemiquinones named as SQNf (fast relaxing) and SQNs (slow relaxing) species [62]. The amplitude [51] and the signal shape [64] seen at given temperature and microwave power is dependent on Delta µH+: the more tight coupling is the higher SQNf signal is seen. In tightly coupled particles, the splitting of the gz line of the N-2 signal can be observed under conditions where the reverse electron transfer occurs, and this splitting correlates with the amplitude of the SQNf signal [61, 62, 64]. The N-2 gz-line splitting magnitude and properties of SQNf suggest the distance between the center N-2 and rapidly relaxing ubisemiquinone of 8-10 Å. It should be noted that the same split of the gz component has been interpreted by Albracht's group as to suggest spin-spin interaction between two N-2 clusters located in the TYKY-subunit (see table) [61].
So far, only two groups have been involved in studies on Complex I-associated ubisemiquinones. Because the membranous preparations derived from prokaryotes are uncoupled (except for sub-bacterial particles of P. denitrificans [65, 66]), no data are available at present on the presence of semiquinones in bacterial or N. crassa Complexes I.
What subunit is ubiquinone bound to? This question cannot be answered yet, although a number of indirect experimental data suggest some speculative proposals. It is generally accepted that several hydrophobic inhibitors (rotenone, piericidine A, piridaben, and others) compete with ubiquinone for binding at the specific site(s) [67, 68]. Photoreactive analogs of rotenone label the 30 kD subunit of mammalian Complex I (ND1 in nomenclature for bovine heart enzyme, NUO-H for prokaryotic enzyme). This subunit is hydrophobic; it contains 8 transmembrane helixes and 4 evolutionarily conserved hydrophilic regions exposed to the mitochondrial matrix (or to the bacterial cytoplasm) [70]. On the other hand, several mutations in the hydrophilic NUO-D subunit containing no transmembrane helixes (homologous to 49 IP subunit of the mitochondrial Complex I) result in a decrease of rotenone- or piericidine A-sensitivity of the enzyme [71, 72]. Recently, piridaben (a tightly-binding inhibitor) was shown to label the PSST-subunit of the mitochondrial Complex I [73] (20 kD nuclear-encoded subunit, NUO-B homolog in prokaryotes, see table). It has been reported many years ago that the small subunit (~15 kD) found in the IP fraction of mammalian Complex I is capable of specific binding of ubiquinone [74]. Unfortunately, no further progress in studies on this small subunit has been reported.
One possible way to account for these apparently contradictory findings is to suggest that relatively hydrophilic “head” of ubiquinone is located in the intersubunit contact area between 49 IP and 20 kD PSST (note direct interaction between SQNf and the N-2 center) and its hydrophobic “tail” is bound to the ND1 subunit. Unfortunately, an identification of the specific quinone-binding motifs in the subunit amino acid sequences is still at a primitive stage [75].
ELECTROCHEMICAL COUPLING
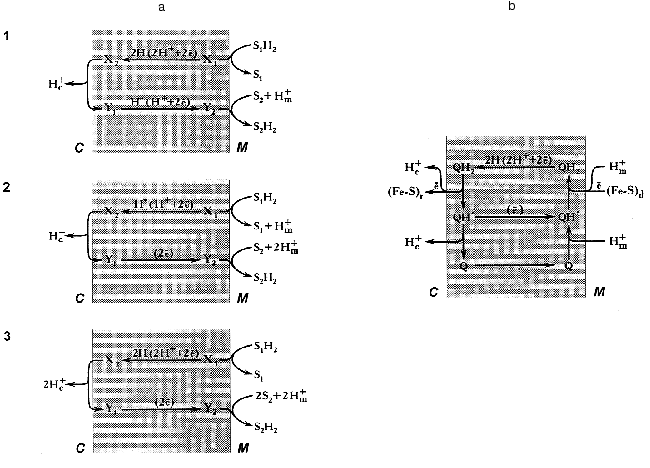
Since this essay is addressed not only to readers who are specialized in bioenergetics, it seems worthwhile to outline briefly some basic concepts of Mitchell's theory on redox-driven proton translocation mechanisms. An earlier hypothesis by Lundegarth suggested a linear transmembrane arrangement of the respiratory chain in such a way that protons derived from a substrate are left on one side of the membrane whereas electrons travel to the other side, and oxygen reduction is accompanied by proton consumption. In contrast, to explain the presence of more than one coupling site in the respiratory chain, Mitchell suggested a proton-translocating loop mechanism [1] (Fig. 2a).
Two requirements should be fulfilled for the operation of any energy-transducing complex according the proton-translocation loop mechanism. First, the complex either must have “multiple” redox components to carry electron and/or hydrogen atom at long distance (~50 Å), or the redox component(s) should be able to diffuse across the membrane. Second, the respiratory complex must have different types of redox components: for example, electron carrier for the lower part of the loop and hydrogen atom carrier for its upper part (scheme 3, Fig. 2a) [Footnote: Here, we do not discuss the so-called “vectorial Bohr effect mechanism” where the redox component is located within the membrane and two wells provide the specific pathways for the protons to be consumed and released at different sides of the coupling membrane [76, 77]. For this type of arrangement the readers are referred to the cytochrome c oxidase coupling mechanism [78]]. A set of the redox components as described in the previous sections meets both requirements: indeed, the iron-sulfur clusters are the electron carriers, FMN is capable to transferring hydrogen atoms, and the ubiquinone/ubisemiquinone couple is able to transfer electrons, hydrogen atoms, and hydride anion. The efficiency of the redox loop mechanism (the number of protons translocated per two electrons transferred) depends on the nature of the particular components participating in the transmembranous transfer of the redox equivalents. For example, for mechanisms 1 (hydrogen atoms move from right to left and hydride anions move from left to right) and 2 (hydride anion moves from right to left and two electrons move from left to right, Fig. 2a) the stoichiometric coefficient n (H+/2e) are equal to 1, and for mechanism 3 (hydrogen atoms move from right to left and electrons move from left to right) n is equal to 2. The latter value is maximal for any single loop mechanism. On the other hand, it is clear that the value of n (if known) makes some restrictions for particular coupling mechanism to be suggested.
Different value of n (1-5) for Complex I have been reported in earlier studies [79]. Recently the value of 4 (pH 8.0) has been determined by our group for the NADH:ubiquinone reductase region of the mammalian respiratory chain [80]. It should be noted that n may depend on pH within the region where protonation/deprotonation of the electron carrier is changing. In any case, a mechanism which gives n greater than 2 cannot be described in terms of a single redox-loop proton translocation. This is also true for the coupling mechanism as operates in Complex III, and the so-called “Q-cycle” mechanism has been proposed [81, 82] (Fig. 2b). A brief account of the Q-cycle seems to be relevant to the present discussion because its basic ideas have been accommodated for almost all hypothetical proposals on the mechanism of proton translocation in Complex I (see [48] for review). In fact, the Q-cycle is a double-loop mechanism (Fig. 2a). It gives overall stoichiometric coefficient n equal to 4 when ubiquinol is oxidized by Complex III coupled with donation of electrons from Complex I or Complex II. Recent resolution of the atomic structure of Complex III showed that the space arrangement of its redox components nicely corresponds to what would be expected for the “Q-cycle” mechanism [83].Fig. 2. a) Three types of mechanisms for vectorial proton translocation from matrix (M) to intermembrane space (C) driven by the oxidoreductase reaction: S1H2 + S2 --> S2H2 + S1 in which both substrate (S1H2 and S2) interact with the enzyme active site exposed to matrix. X and Y, redox components that form a “loop” according to Mitchell [1]. b) Electrochemical coupling in ubiquinol:cytochrome c reductase (Complex III) according to the “Q-cycle” mechanism [81, 82]. Q, QH*, and QH2, oxidized, semiquinone and reduced ubiquinone, respectively. (Fe-S)d and (Fe-S)r, the terminal iron-sulfur centers of dehydrogenases (Complex I and Complex II) and iron-sulfur Rieske protein, respectively. Electrons from (Fe-S)r are transferred to cytochrome c located in the intermembrane space. Two cytochrome b hemes transfer electrons between two ubisemiquinones (not shown).
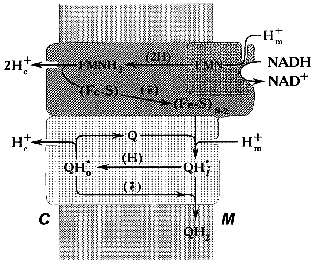
In our working hypothesis on the mechanism of electrochemical coupling in Complex I published about ten years ago [52], we accommodated Mitchell's ideas to explain the experimental data on the Complex I-associated ubisemiquinone. This hypothesis is discussed in some detail below. Note should be made that the major point of our proposal was to emphasize the role of ubiquinone in the proton-translocating activity of Complex I (Fig. 3). According to our scheme, oxidation of FMNH2 by bulk ubiquinone is coupled to proton translocation. The iron-sulfur centers N-3 and N-4 are involved in oxidation of FMNH2 by the N-2 cluster localized between the hydrophobic and hydrophilic parts of the “boot”. Two protons are translocated from matrix to intermembrane space per pair of electrons transferred from FMNH2 to N-2. It is assumed that two types of FMN binding exist, that is, oxidized flavin can be accessible for protons from the matrix and the reduced cofactor can be deprotonated to release protons into the intermembrane space. The iron-sulfur cluster N-2 is an electron donor for the bound ubiquinone and its one-electron reduction is accompanied by uptake of one proton from the matrix to form electroneutral ubisemiquinone Q*H+ at site i (SQNf). Neutral ubisemiquinone transfers one electron and one proton to the ubiquinone bound at site o, thus transfer of two electrons from N-2 to the bound ubiquinone results in a formation of two protonated electroneutral ubisemiquinones (SQNf and SQNs) located at sites o and i, respectively. The next step which we called vectorial dismutation is Delta µH+ generation: Q*H+i (SQNf) oxidizes Q*oH+ (SQNs) in an electrogenic reaction (Delta iota generation) leaving proton bound to SQNs outside and taking a proton from the matrix space to SQNf. This dismutation results in oxidized (at site o) and reduced (at site i) ubiquinone molecules that are exchangeable with bulk ubiquinone. This exchange is prevented by the specific inhibitors rotenone and piericidine. The vectorial dismutation results in translocation of one proton per two electrons needed for two-step full reduction of Q to QH2. To make the system operate as a proton pump, the reduction of ubisemiquinone by N-2 at site i should be prevented. Thus, it was postulated that the formation of SQNf results in the conformational state of N-2 which is not able to transfer an electron to Q*oH+ bound at the i site. The same problem arises in Complex III operating in Q-cycle mechanism (see Fig. 2b): if ubisemiquinone formed at the outer side of the coupling membrane would be oxidized by cytochrome c, this would result in a decrease of energetic efficiency of the overall reaction. This problem is solved by placing a Rieske iron-sulfur center on a specially designed flexible arm [83]. Our scheme was able to explain why the rapidly relaxing SQNf is seen under the steady-state only in tightly coupled preparations. Indeed, electrogenic vectorial dismutation is expected to be prevented by Delta µH+ and it proceeds rapidly in uncoupled preparations. Also, our hypothesis predicted that two ubisemiquinone species located at different distance from N-2 with different relaxation time must be present. Two forms of ubisemiquinone (SQNf and SQNs) have indeed been observed [62, 64].
Recently, new data have been obtained that seem difficult to accommodate in our original scheme. The model as depicted in Fig. 3 gives the overall stoichiometry of 3 H+ per 2 electrons, whereas the experimental n value of 4 has been determined [80]. This discrepancy is easy to overcome in “paper biochemistry” by postulating the vectorial proton translocation coupled with oxidation-reduction of N-2 (note that the midpoint redox potential of N-2 is pH dependent). The structural basis for the functioning of the N-2-containing subunit as a proton pump may be analogous or even homologous to the proton- and hydrogen-conducting pathways found in the structure of hydrogenase from anaerobic Disulfovibrio gigas [84]. For this enzyme, it has been demonstrated that deuterium exchange of several amino-acid residues buried in the protein interior where the iron-cluster is located is permitted in the reduced protein and prevented when the cluster is oxidized [85]. Interestingly, alpha-, beta- and gamma-subunits of hydrogenase from the lithotrophic anaerobe Alcaligenes eutropus are highly homologous to FP and IP subunits of the mitochondrial Complex I [4, 86]. Space does not permit further discussion of this very interesting problem.Fig. 3. Hypothetical electrochemical coupling in Complex I [52]. For the details, see text.
The other “news” recently reported by our group is that Complex I is fully capable of proton translocation with the same stoichiometry (n = 4) in the presence of rotenone or being in the de-activated state [80, 87], i.e., under the conditions where the electron transfer from N-2 to bulk ubiquinone is blocked. This finding, which contradicts the generally accepted view on the rotenone-sensitivity of the first coupling site, can also be explained by our scheme. It can be proposed that relatively hydrophilic ubiquinone-1 (rotenone-insensitive fraction of NADH-Q1 reductase) accepts electrons from ubiquinol formed at site i located at the outer side of inside-out submitochondrial particles.
Space limitation does not permit to discuss here the other models of electrochemical coupling in Complex I (more than 10) that have been published in the current literature. For those schemes readers are referred to recently published review [48]. One of the newest models merits some comments. A mechanism for coupling in Complex I has been recently proposed by Dutton and his associates, in which all steps of electrochemical coupling take place at bound ubiquinone oxidoreduction level [88]. This model, which is formulated following the traditional Mitchellian view, is in fact the proton-translocating Q-cycle as suggested for Complex III [81, 82] but operating in opposite direction with ubiquinone (instead of ubiquinol) and ubiquinol (instead of ubiquinone) as the substrate and product, respectively, and translocating protons from matrix to outside of mitochondria. All the redox components (FMN and the iron-sulfur clusters) are assumed to be an electron injector which provides highly negative redox potential at the first component participating in the coupling mechanism [88]. My major objection to this model is that it can hardly explain Delta µH+-dependent reverse electron transfer from ubiquinol to NAD+, which accepts electrons at the hydrophilic part of the enzyme (most likely from FMN), a reaction that is energy-dependent and that proceeds at the rate of about one-fourth of the NADH-ubiquinone reductase activity [7]. Note should be made here that according to our hypothesis the electron pathways for the forward and reverse reaction within Complex I are not identical [22, 89, 90].
COMPLEX I AS A CONFORMATIONALLY COUPLED PROTON PUMP
The remarkable feature of Mitchell's coupling mechanism is the precise indication of their particular participants. It is appropriate to note that the first postulate in the original version of his chemiosmotic theory was the presence of the membrane-bound ATPases “... and their normal function is to couple reversibly the translocation of protons across the membrane to the flow of anhydro-bound equivalents between water and the couple ATP/(ADP + Pi)” [1]. In other words, for a long time Mitchell advocated a mechanism where nucleotide and phosphate are the immediate donors/acceptors of protons which are coming/leaving to/from the ATPase active site from/to the specific proton channel Fo [91]. Further numerous studies on FoF1 have shown this mechanism is not operating and the chemical events (ATP formation) at the enzyme active site is energetically coupled with the proton flow by long distance conformational change (see series of recent reviews published in a special issue of Journal of Bioenergetics and Biomembranes [92]). Now most scholars in the field agree that the “conformational coupling mechanism” originally proposed by P. Boyer [93] is valid in FoF1-ATP synthase. This mechanism is operating as a rotation of one “long” subunit (gamma) interacting by one terminal with the nucleotide-binding active sites whereas the other end is connected to the proton-conducting ring of 9-12 c subunits [94, 95]. Although I am not convinced that the rotary mechanism of ATP synthesis has been conclusively proven, the “conformational coupling mechanisms” seem very likely to operate in FoF1-ATP synthase and also in V-type (vacuolar proton-translocating ATPase [96]) and in P-type (Na+/K+-ATPase [97], sarcoplasmic reticulum Ca2+-ATPase [98]) ATPases.
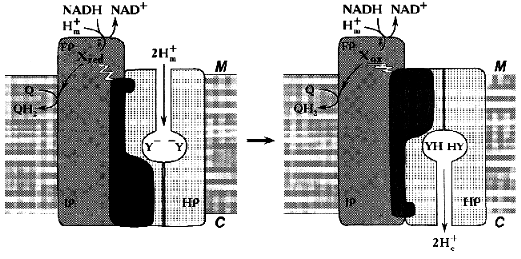
Similar to what is known for the proton- or other cation-translocating ATPases, the nucleotide-binding site(s) and most (if not all) redox components in Complex I are likely to be located distantly from the coupling membrane. Hatefi and his associates [40] were the first to suggest the possibility of Boyer's “alternating binding change mechanism” to operate in Complex I. Our attempts to find tightly bound nucleotides in Complex I have failed (N. Zakharova and A. Vinogradov, unpublished observation). However, the absence of tightly bound substrates/products in Complex I does not exclude the hypothesis that can be formulated as follows: none of the enzyme redox components is directly involved in proton translocation and the enzyme operates as a redox-coupled conformationally driven pump (Fig. 4). The oxidation/reduction transitions of the components located in the hydrophilic part of the enzyme are accompanied by conformational rearrangements [37, 40] that are further transformed to the membrane-associated hydrophobic part. The membrane-embedded part (HP) serves as a proton channel, whereas some hydrophilic subunits containing no redox components serve as an energy-transducing mechanical part of the machine. Such a mechanism easily explains high stoichiometric coefficient for proton translocation: the reduction of one or more components is coupled with occlusion of two (or even more) protons in the channel and the subsequent oxidation results in release of these protons on the other side of the coupling membrane. Such a mechanism is especially attractive for Na+-translocating NADH:quinone oxidoreductases [99, 100] which contain flavins and iron-sulfur centers and which are genetically related [100] or unrelated [99] to the mitochondrial Complex I. For these enzymes, elegant Mitchellian type models are hard to accept because of unlikely chemistry. I believe that when the structure of Complex I will be visualized, all the redox components including the ubiquinone “head” binding site(s) may well be found far away from the membrane.
It can hardly be denied that this model is as hypothetical as all other models proposed previously [48] including ours [52]. It may, however, be helpful for planning further experiments aimed to disprove the proposed mechanism.Fig. 4. Conformational coupling in Complex I. All redox reactions take place in the hydrophilic part of the enzyme. The reduction of one or several redox component(s) (X) is accompanied by occlusion of protons (protonation of Y-groups) in the specific proton channel formed by HP peptides. Oxidation of Xred results in release of occluded protons into the intermembrane space C.
I am grateful to all my colleagues, the coauthors of our papers [20, 22, 26, 37, 41, 50, 51, 57, 62, 63, 80, 87, 89, 90] who constantly inspire my guess-work on the mechanism of Complex I. Thanks are due to Dr. F. Kasparinsky for his help in preparation of graphic materials.
The work done in my laboratory was partially supported by the Russian Foundation for Basic Research (grant 99-04-48082), by the Program “Leading Schools in Science” (grant 00-15-97798), and by The Royal Swedish Academy of Science (grant 12557).
REFERENCES
1.Mitchell, P. (1966) Chemiosmotic Coupling
Oxidative and Photosynthetic Pphosphorylation, Glynn Research
Ltd.
2.Mitchell, P. (1979) David Keilin's Respiratory
Chain Concept and Its Chemiosmotic Consequences, The Nobel
Foundation.
3.Fearnley, I. M., and Walker, J. E. (1992)
Biochim. Biophys. Acta, 1140, 105-134.
4.Walker, J. E. (1992) Q. Rev. Biophys.,
25, 253-324.
5.Friedrich, T., and Weiss, H. (1997) J. Theor.
Biol., 187, 529-541.
6.Friedrich, T., and Scheide, D. (2000) FEBS
Lett., 479, 1-5.
7.Vinogradov, A. D. (1998) Biochim. Biophys.
Acta, 1364, 169-185.
8.Ohnishi, T. (1979) in Membrane Proteins in
Energy Transduction (Capaldi, R. A., ed.) Marcel Dekker, Inc., New
York, pp. 1-87.
9.Sled, V. D., Friedrich, T., Leif, H., Weiss, H.,
Meinhardt, S. W., Fukumori, Y., Caihoun, M. W., Gennis, R. B., and
Ohnishi, T. (1993) J. Bioenerg. Biomembr., 25,
347-355.
10.Robinson, B. H. (1998) Biochim. Biophys.
Acta, 1364, 271-286.
11.Schapira, A. H. V. (1998) Biochim. Biophys.
Acta, 1366, 225-233.
12.Chomyn, A., Mariottini, P., Cleeter, M. W. J.,
Ragan, C. I., Matsuno-Yagi, A., Hatefi, Y., Doolittle, R. F., and
Attardi, G. (1985) Nature, 314, 592-597.
13.Yagi, T., Yano, T., Di Bernardo, S., and
Matsuno-Yagi, A. (1998) Biochim. Biophys. Acta, 1364,
125-133.
14.Dupuis, A., Chevalett, M., Darrouzet, E.,
Duborjal, H., Lunardi, J., and Issartel, J. P. (1998) Biochim.
Biophys. Acta, 1364, 147-165.
15.Tuschen, G., Sackmann, U., Nehls, U., Haiker, H.,
Buse, G., and Weiss, H. (1990) J. Mol. Biol., 213,
845-857.
16.Hofhaus, G., Weiss, H., and Leonard, K. (1991)
J. Mol. Biol., 221, 1027-1043.
17.Guénebaut, V., Schlitt, A., Weiss, H.,
Leonard, K., and Friedrich, T. (1998) J. Mol. Biol., 276,
105-112.
18.Walker, J. E., Arizmendi, J. M., Dupuis, A.,
Fearnley, I. M., Finel, M., Medd, S. M., Pilkington, S. J., Runswick,
M. J., and Skehel, J. M. (1992) J. Mol. Biol., 226,
1051-1072.
19.Ernster, L., Dallner, G., and Azzone, G. F.
(1963) J. Biol. Chem., 238, 1124-1131.
20.Grivennikova, V. G., Maklashina, E. O.,
Gavrikova, E. V., and Vinogradov, A. D. (1997) Biochim. Biophys.
Acta, 1319, 223-232.
21.Jeng, M., Holl, C., Crane, F. L., Takahashi, M.,
Tamura, S., and Folkers, K. (1968) Biochemistry, 7,
1311-1322.
22.Ushakova, A. V., Grivennikova, V. G., Ohnishi,
T., and Vinogradov, A. D. (1999) Biochim. Biophys. Acta,
1409, 143-153.
23.Degli Esposti, M. (1998) Biochim. Biophys.
Acta, 1364, 222-235.
24.Lümmen, P. (1998) Biochim. Biophys.
Acta, 1364, 287-296.
25.Hatefi, Y., and Rieske, J. S. (1967) Meth.
Enzymol., 10, 235-239.
26.Vinogradov, A. D., Gavrikova, E. V.,
Grivennikova, V. G., Zharova, T., and Zakharova, N. V. (1999)
Biochemistry (Moscow), 64, 1219-1229.
27.Galante, Y. M., and Hatefi, Y. (1978) Meth.
Enzymol., 53, 15-21.
28.Paech, C., Friend, A., and Singer, T. P. (1982)
Biochem. J., 203, 477-481.
29.Finel, M., Skehel, M., Albracht, S. P. J.,
Fearnley, I. M., and Walker, J. E. (1992) Biochemistry,
31, 11425-11434.
30.Brink, J., Hovmöller, S., Ragan, C. I.,
Cleeter, M. W. J., Boekema, E. J., and van Bruggen, E. F. J. (1987)
Eur. J. Biochem., 166, 287-294.
31.Albracht, S. P. J. (1982) in Flavins and
Flavoproteins (Massey, V., and Williams, C. H., eds.)
Elsevier/North-Holland Inc., pp. 759-762.
32.Van Belzen, R., van Gaalen, M. C. M., Cupyers, P.
A., and Albracht, S. P. J. (1990) Biochim. Biophys. Acta,
1017, 152-159.
33.Van Belzen, R., De Jong, A. M. P., and Albracht,
S. P. J. (1992) Eur. J. Biochem., 209, 1019-1022.
34.Ragan, C. I. (1976) Biochim. Biophys.
Acta, 456, 249-290.
35.Galante, Y. M., and Hatefi, Y. (1979) Arch.
Biochem. Biophys., 192, 559-568.
36.Belogrudov, G., and Hatefi, Y. (1994)
Biochemistry, 33, 4571-4576.
37.Sled, V. D., and Vinogradov, A. D. (1993)
Biochim. Biophys. Acta, 1143, 199-203.
38.Clark, W. M. (1960) Oxidative Reduction
Potentials of Organic Systems, Williams and Wilkins, Baltimore,
USA.
39.Mayhew, S. G. (1999) Eur. J. Biochem.,
265, 698-702.
40.Yamaguchi, M., Belogrudov, G. I., and Hatefi, Y.
(1998) J. Biol. Chem., 273, 8094-8098.
41.Gavrikova, E. V., Grivennikova, V. G., Sled, V.
D., Ohnishi, T., and Vinogradov, A. D. (1995) Biochim. Biophys.
Acta, 1230, 23-30.
42.Sled, V. D., Rudnitzky, N. I., Hatefi, Y., and
Ohnishi, T. (1994) Biochemistry, 33, 10069-10075.
43.Massey, V., and Palmer, G. (1966)
Biochemistry, 5, 3181-3189.
44.Fecke, W., Sled, V. D., Ohnishi, T., and Weiss,
H. (1994) Eur. J. Biochem., 220, 551-558.
45.Ohnishi, T. (1981) in Mitochondria and
Microsomes (Lee, C. P., Schatz, G., and Dallner, G., eds.)
Addison-Wesley Publishing Company, Inc., Advanced Book Program/World
Science Division, pp. 191-216.
46.Ohnishi, T. (1975) Biochim. Biophys. Acta,
387, 475-490.
47.Kröger, A., and Unden, G. (1985) in
Coenzyme Q (Lenaz, G., ed.) Wiley, New York, pp. 285-300.
48.Brandt, U. (1996) Biochim. Biophys. Acta,
1318, 79-91.
49.Ohnishi, T. (1976) Eur. J. Biochem.,
64, 91-98.
50.Burbaev, D. Sh., Moroz, I. A., Kotlyar, A. B.,
Sled, V. D., and Vinogradov, A. D. (1989) FEBS Lett.,
254, 1, 47-51.
51.Kotlyar, A. B., Sled, V. D., Burbaev, D. Sh.,
Moroz, I. A., and Vinogradov, A. D. (1990) FEBS Lett.,
264, 17-20.
52.Vinogradov, A. D. (1993) J. Bioenerg.
Biomembr., 25, 367-375.
53.Meinhardt, S. W., Kula, T., Yagi, T., Lillich,
T., and Ohnishi, T. (1987) J. Biol. Chem., 262,
9147-9153.
54.Meinhardt, S. W., Matsushita, K., Kaback, H. R.,
and Ohnishi, T. (1989) Biochemistry, 28, 2153-2160.
55.Krishnamoorthy, G., and Hinkle, P. (1988) J.
Biol. Chem., 263, 17566-17575.
56.Suzuki, H., and King, T. E. (1983) J. Biol.
Chem., 258, 352-358.
57.Kotlyar, A. B., and Vinogradov, A. D. (1990)
Biochim. Biophys. Acta, 1019, 151-158.
58.Albracht, S. P. J., van Belzen, R., and De Jong,
A. M. P. (1991) Biol. Chem. Hoppe Seyler, 372, 547.
59.De Jong, A. M. P., and Albracht, S. P. J. (1994)
Eur. J. Biochem., 222, 975-982.
60.Van Belzen, R., Kotlyar, A. B., Moon, N., Dunham,
W. R., and Albracht, S. P. J. (1997) Biochemistry, 36,
886-893.
61.Albracht, S. P. J., and De Jong, A. M. P. (1997)
Biochim. Biophys. Acta, 1318, 92-106.
62.Vinogradov, A. D., Sled, V. D., Burbaev, D. S.,
Grivennikova, V. G., Moroz, I. A., and Ohnishi, T. (1995) FEBS
Lett., 370, 83-87.
63.Ohnishi, T., Magnitsky, S., Toulokhonova, L.,
Yano, T., Yagi, T., Burbaev, D. S., and Vinogradov, A. D. (1999)
Biochem. Soc. Trans., 27, 586-591.
64.Ohnishi, T., Sled, V. D., Yano, T., Yagi, T.,
Burbaev, D. S., and Vinogradov, A. D. (1998) Biochim. Biophys.
Acta, 1365, 301-308.
65.John, P., and Whatley, F. R. (1977) Biochim.
Biophys. Acta, 463, 129-153.
66.Kotlyar, A. B., Albracht, S. P. J., and van
Spanning, R. J. M. (1998) Biochim. Biophys. Acta, 1365,
53-59.
67.Okun, J. G., Lummen, P., and Brandt, U. (1999)
J. Biol. Chem., 274, 2625-2630.
68.Friedrich, T., van Heek, P., Lief, H., Ohnishi,
T., Forche, E., Kunze, B., Jansen, R., Trowitzsch-Kienast, W.,
Höfle, G., Reichenbah, H., and Weiss, H. (1994) Eur. J.
Biochem., 219, 691-698.
69.Earley, F. G. P., and Ragan, C. I. (1984)
Biochem. J., 224, 525-534.
70.Roth, R., and Hägerhäll, C. (2001)
Biochim. Biophys. Acta, 1504, 352-362.
71.Darrouzet, E., Issartel, J. B., Lunardi, J., and
Dupuis, A. (1998) FEBS Lett., 431, 34-38.
72.Prieur, I., Lunardi, J., and Dupuis, A. (2001)
Biochim. Biophys. Acta, 1504, 173-178.
73.Schuler, F., Yano, T., Di Bernardo, S., Yagi, T.,
Yankovskaya, V., Singer, T. P., and Casida, J. E. (1999) Proc. Natl.
Acad. Sci. USA, 96, 4149-4153.
74.Suzuki, H., and Ozawa, T. (1986) Biochem.
Biophys. Res. Commun., 138, 1237-1242.
75.Fisher, N., and Rich, P. (2000) J. Mol.
Biol., 296, 1153-1162.
76.Papa, S., Lorusso, M., and Capitanio, N. (1994)
J. Bioenerg. Biomembr., 26, 609-618.
77.Skulachev, V. P. (1975) Curr. Top.
Bioenerg., 4, 127-185.
78.Babcock, G. T., and Wikström, M. (1992)
Nature, 356, 301-308.
79.Hinkle, P. C. (1981) in Chemiosmotic Proton
Circuits in Biological Membranes (Skulachev, V. P., and Hinkle, P.
C., eds.) Addison-Wesley Publ. Inc., Massachusetts, pp. 49-58.
80.Galkin, A. S., Grivennikova, V. G., and
Vinogradov, A. D. (1999) FEBS Lett., 451, 157-161.
81.Mitchell, P. (1975) FEBS Lett., 56,
1-6.
82.Mitchell, P. (1976) J. Theor. Biol.,
2, 327-367.
83.Zhang, Z., Huang, L., Shulmeister, V. M.,
Young-In Chi, Kim, K. K., Li-Wei Hung, Crofts, A. R., Berry, E. A., and
Sung-Hou Kim (1998) Nature, 392, 677-684.
84.Volbeda, A., Charon, M.-H., Piras, C.,
Hatchikian, E. C., Frey, M., and Fontecilla-Camps, J. C. (1995)
Nature, 373, 580-587.
85.Chapman, A., Cammack, R., Hatchikian, C. E.,
McCracken, J., and Peisach, J. (1988) FEBS Lett., 242,
134-138.
86.Tran-Betcke, A., Warnecke, U., Böcker, C.,
Zabarosch, C., and Friedrich, B. (1990) J. Bacteriol.,
172, 2920-2929.
87.Galkin, A. S., Grivennikova, V. G., and
Vinogradov, A. D. (2001) Biochemistry (Moscow), 66,
435-443.
88.Dutton, P. L., Moser, C. C., Sled, V. D., Daldal,
F., and Ohnishi, T. (1998) Biochim. Biophys. Acta, 1364,
245-257.
89.Zharova, T. V., and Vinogradov, A. D. (1997)
Biochim. Biophys. Acta, 1320, 256-264.
90.Zakharova, N. V., Zharova, T. V., and Vinogradov,
A. D. (1999) FEBS Lett., 444, 211-216.
91.Mitchell, P. (1974) FEBS Lett., 43,
189-194.
92.Pedersen, P. L. (ed.) (1996) J. Bioenerg.
Biomembr., 28, 398-451.
93.Boyer, P. D. (1975) FEBS Lett., 58,
1-6.
94.Tsunoda, S. P., Aggeler, R., Yoshida, M., and
Capaldi, R. A. (2001) Proc. Natl. Acad. Sci. USA, 98,
898-902.
95.Sambongi, Y., Ueda, I., Wada, Y., and Futai, M.
(2000) J. Bioenerg. Biomembr., 32, 441-448.
96.Forgac, M. (2000) J. Exp. Biol.,
203, 61-70.
97.Jorgensen, P. L., Nielsen, J. M., Rasnussen, J.
H., and Pedersen, P. A. (1998) Biochim. Biophys. Acta,
1365, 65-70.
98.Toyochima, C., Nakasako, M., Nomura, H., and
Ogawa, H. (2000) Nature, 405, 647-655.
99.Unemoto, T., and Hayashi, M. (1993) J.
Bioenerg. Biomembr., 25, 385-391.
100.Krebs, W., Steuber, J., Gemperli, A. C., and
Dimroth, P. (1999) Mol. Microbiol., 33, 590-598.