Study of the Properties of Phosphorylating D-Glyceraldehyde-3-phosphate Dehydrogenase
N. K. Nagradova
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: serene@cityline.ru
Received April 16, 2001; Revision received June 10, 2001
The properties of the active center of phosphorylating D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) are considered with emphasis on the structure of anion-binding sites and their role in catalysis. The results of studies on the molecular mechanism of the effect of NAD+ on the enzyme conformation are discussed. Experimental evidence is presented supporting the idea that negative cooperativity of NAD+ binding and half-of-the-sites reactivity exhibited by GAPDH are generated by different mechanisms. Data obtained with rabbit muscle and Escherichia coli GAPDH point to preexisting asymmetry in these tetramers. Structural determinants that can control the transition of the tetramer from the symmetric to the asymmetric state were found.
KEY WORDS: D-glyceraldehyde-3-phosphate dehydrogenase, catalytic mechanism, active center, domains, half-of-the-sites reactivity, preexisting asymmetry
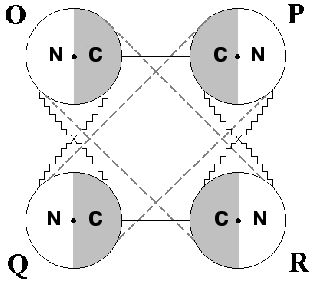
Phosphorylating D-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an enzyme that has long attracted the attention of many investigators. The reason is that in spite of considerable progress achieved in solving the structure of this homotetramer and in elucidating the fundamentals of its molecular mechanism of catalysis, a fair number of questions about it still remains unanswered. A major question has to do with the structural basis underlying the stabilization of different conformational states of the oligomer. To understand this, one has to elucidate the contribution of separate types of intramolecular interactions, primarily interdomain interactions, to such stabilization. Figure 1 demonstrates this idea. Each subunit of the tetrameric GAPDH molecule is composed of two domains, an NAD+-binding domain and a catalytic domain. The location of the active center in the interdomain area provides for a direct relationship between its conformational state and the way the NAD+-binding and the catalytic domains interact within each monomer.
As seen in Fig. 1, intersubunit interactions via NAD+-binding and catalytic domains also exist between adjacent monomers; only between O and P subunits and between Q and R subunits are the interactions formed via their catalytic domains alone [1-3]. This complex web of interdomain interactions creates the structural basis for the functional interplay between subunits within the oligomer, the most important manifestations thereof being their cooperativity in NAD+ binding and the so-called half-of-the-sites reactivity (a non-equivalence of active centers upon interaction with some substrate analogs or inhibitors).Fig. 1. Schematic representation of the tetrameric structure of phosphorylating D-glyceraldehyde-3-phosphate dehydrogenase. N is the nucleotide-binding domain; C is the catalytic domain. Contacts between subunits are represented by lines. Dots mark the location of active centers.
The molecular mechanism of these effects has been the subject of numerous studies regarded today as classical works of modern enzymology ([4-15] and many others). They have led to the accumulation of a large body of knowledge and to the development of several alternative models, but a definitive answer continues to evade researchers. This seems to be due largely to insufficiency of our knowledge on the conformational states of the active center during different catalytic steps and on the factors that can affect those states. Available information on the three-dimensional structure of the enzyme is still incomplete (no high-resolution X-ray crystallography data have been obtained yet for the tertiary enzyme*NAD+*substrate analog complex. At the same time, progressive refining of this method and the widening range of objects studied provide deeper insights into the problem. The development of highly sensitive methods which detect changes in protein conformation (e.g., differential scanning calorimetry) offers the possibility to evaluate the contribution of individual types of intramolecular interactions to the stabilization of one or another conformational state.
Part of our research covered in this review was aimed at obtaining new information on the properties of the active center of GAPDH in the hope of better understanding the molecular mechanisms that determineits functioning. Another problem discussed in this review is related to the study of different manifestations of active center cooperativity within the tetrameric enzyme molecule and of the interrelationship between their respective mechanisms. One of our main tasks in writing this review was to discuss the relevant results in their entirety against the backdrop of present-day perceptions as to the structural and functional properties of GAPDH.
ROLE OF Arg231 IN THE CATALYTIC MECHANISM OF GAPDH
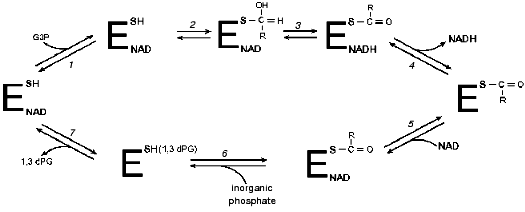
By the time X-ray crystallography data first appeared [16], the principal steps of the GAPDH-catalyzed reaction had already been established (see Fig. 2). Solid data existed documenting the character of functional groups involved in catalysis. The X-ray crystallographic analysis confirmed the participation of Cys149 as a nucleophilic acyl group acceptor and of His176 as a general base that accepts a proton upon substrate oxidation. Quite unexpectedly, however, an arginine residue (Arg231) was detected in proximity to Cys149 [16]. By analogy with lactate dehydrogenase, whose active center also contained an arginine residue, the authors (M. Rossmann et al.) suggested a similar function for the arginine residues: stabilization of negative charges developing at various stages of the dehydrogenase reactions [18]. It was hypothesized that Arg231 may participate in destabilization of the Michaelis complex [19].
The first experimental studies aimed at elucidating the functional role of the arginine residue in the active center of GAPDH were performed in our laboratory with enzymes isolated from rat and rabbit skeletal muscle and from baker's yeast [20-28]. It was found that 2,3-butanedione specifically modifies one arginine residue per subunit of the tetrameric rabbit muscle GAPDH, and, as was shown more recently, also of the Escherichia coli (E. coli)and Bacillus stearothermophilus (B. stearothermophilus)GAPDHs. The identification of this residue in the primary structure of GAPDHs from different sources [29, 30] showed that the modification affected Arg231, a residue always present in the structures of all GAPDHs studied so far. Its selective reactivity may be determined, among other factors, by peculiarities of the active center environment, probably responsible for the lowering of the apparent pKa of this residue to 9.0 [28].Fig. 2. Kinetic mechanism of GAPDH proposed by Segal and Boyer [17]. Stages: 1) binding of glyceraldehyde-3-phosphate (G3P) with holoenzyme; 2) formation of a thiohemiacetal; 3) hydride ion transfer; 4) release of NADH; 5) binding of NAD+ with acyl-enzyme; 6) phosphorolysis; 7) release of 1,3-bisphosphoglycerate (1,3-dPG). R = CH(OH)CH2OPO32-.
Modification of Arg231 resulted in a sharp (up to 95%) decrease in the rate of catalysis. In studies of the functional role of the arginine, we considered the possibility that it may participate in the binding of anions, in particular, of the phosphate group of the substrate, glyceraldehyde-3-phosphate (3-PGA). The experiments on chemical modification of apo-GAPDH by 2,3-butanedione revealed no effect of 3-PGA on the rate and extent of the modification, leading us to conclude that Arg231 does not participate in the binding of substrate to the apoenzyme. However, we could not rule out the possibility that an anion-binding site comprising Arg231 and responsible for the interaction with 3-PGA is formed only upon NAD+ binding, i.e., exists only in the holoenzyme.
In agreement with this possibility, the rate constant of Arg231 modification increased markedly in the presence of NAD+, indirectly pointing to alterations in the microenvironment of this residue upon transition from apo to holo conformation [21]. Our subsequent experiments demonstrated that 3-PGA effectively protects Arg231 from modification occurring in the presence of NAD+; upon extrapolation of the data to saturating substrate concentrations, complete protection was achieved. Similar results were obtained with GAPDHs isolated from rabbit and rat skeletal muscle and from baker's yeast [22-24]. Using isolated catalytically active subunits of the enzyme stabilized by immobilization on a solid support, we showed that the effect of NAD+ on the reactivity of the arginine residue modified by 2,3-butanedione, as well as the protective effect of substrate are realized even within a separate monomer [25-27]. The overall result of these studies was the suggestion that 3-PGA might bind differently to apo- and holo-GAPDH.
Testing the validity of this concept by direct structural analysis became possible only a few years ago, after an X-ray crystallography study was carried out on the complex of E. coli GAPDH with substrate [31]. To obtain crystals of the covalent adduct apoenzyme-thiohemiacetal, apo-GAPDH crystals were incubated in the presence of 3-PGA and then frozen. The substrate-binding site was found to be located in the catalytic domain and formed by the side-chains of Ser148, Thr150, and Thr208, hydrogen bonded to the oxygen atoms of the 3-PGA phosphate group, and by the main chain nitrogen atoms of Thr150 and Gly209, also forming hydrogen bonds with the above phosphate group. The fact that Arg231 is located somewhat aside from this anion-binding site is in good agreement with a conclusion derived from our own experiments according to which this residue does not participate in the binding of 3-PGA with the apoenzyme.
A few words should be said regarding the current state of our knowledge on the anion-binding sites of the holoenzyme and their role in different steps of catalysis [1, 32]. The two anion-binding sites revealed in the active center of the holo-GAPDH were identified as the inorganic phosphate binding site (Pi, formed by residues Ser148, Thr150, Thr208, and others and discussed above) and the site involved in the binding of the substrate phosphate group (Ps, formed by residues Arg231, Thr179, and Asn181); this site also contains a hydroxy group at the second carbon atom of the nicotinamide ribose of the bound NAD+ molecule. The latter fact is consistent with the idea that the presence of the coenzyme is a prerequisite for the formation of the Ps site [22].
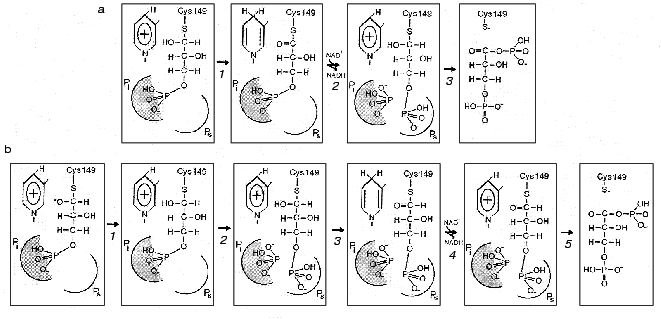
The difficulties encountered in fitting the thiohemiacetal structure into the active center combined with the results of structural analysis of the holoenzyme*glycidol-3-phosphate complex, whose phosphate group appeared to be bound at the Pi site and not at the Ps site, gave reason to suggest a “flipping” of the substrate phosphate group from one site to another upon transition from the oxidative step of the reaction to the deacylation step. Thus, in the course of thiohemiacetal formation and subsequent oxidoreduction the substrate remains bound at the Pi site, then becomes “flipped off” as a result of conformational change induced by the substitution of NAD+ for NADH, and is brought into the Ps site. As this takes place, inorganic phosphate occupies the Pi site, from where it launches a nucleophilic attack on the first carbon atom of the substrate in the phosphorolysis reaction [1]. This mechanism is illustrated in Fig. 3a.
To test the above hypothetical mechanism experimentally, Corbier et al. [30] performed a series of kinetic experiments with B. stearothermophilus GAPDH containing mutations at residues involved in the formation of each of the two anion-binding sites. In the case of the Ps site, Arg231 was replaced by Gly, whereas in the case of the Pi site, Arg195 was mutated. An investigation of the effect of these mutations on the two reaction steps--oxidoreduction, which involves the formation of thiohemiacetal and a hydride ion transfer (Fig. 2, steps 1-3), and phosphorolysis (Fig. 2, step 6)--showed that the replacement of Arg195 by Leu dramatically reduces the rate of oxidoreduction, while the change of Arg231 to Gly has little or no effect on this step of the reaction. On the other hand, the replacement of Arg231 considerably slows down the acyl-enzyme phosphorolysis. In sum, the results obtained were consistent with the hypothetical scheme shown in Fig. 3a where Arg231 plays its role during the conformational isomerization step, which is induced by the substitution of NAD+ for NADH and results in the “flipping” of the substrate phosphate group and also takes part in phosphorolysis.Fig. 3. Two proposed mechanisms of the participation of anion-binding sites in the orientation of substrate at the active site of GAPDH complexed with NAD+ at different steps of catalysis. a: 1) Thiohemiacetal, bound via its phosphate group in the Pi center, is oxidized producing a ternary 3-phosphoglyceroyl-enzyme*NADH complex; 2) subsequent replacement of NADH by NAD+ results in a conformational isomerization of the acyl-enzyme, which is followed by a “flipping” of the phosphate group from the Pi site to the Ps site. The Pi site becomes occupied by inorganic phosphate; 3) inorganic phosphate bound in the Pi site attacks the first carbon atom of the acyl-enzyme to form 1,3-bisphosphoglycerate. b: 1) Formation of thiohemiacetal, occurring with its phosphate group bound in the Pi site, is accelerated owing to the stabilization of the transition state oxyanion by the positive charge of NAD+; 2) the thiohemiacetal phosphate group “flips” to the Ps site due to a change in the geometry of the thiohemiacetal molecule (not reflected in the figure but described by the authors of the scheme [31]). Possibly, the newly vacant Pi is filled by inorganic phosphate. The oxidoreduction (3), nucleotide exchange (4), and phosphorolysis (5) steps proceed with the substrate phosphate group fixed at the Ps site.
Several years earlier we carried out a similar kinetic investigation on rabbit muscle GAPDH modified at Arg231 with 2,3-butanedione [33]. It showed the modification of Arg231 to result in a considerable lowering of the first-order rate constant of the acyl-enzyme*NADH complex formation and markedly affect the rate of phosphorolysis. These data suggested that the functions of the two anion-binding sites in the active center of rabbit muscle GAPDH might somewhat differ from those in the active center of the B. stearothermophilus enzyme. Namely, it seemed possible that in the case of rabbit muscle GAPDH, the Ps site is involved in both the oxidoreduction step, causing the formation of acyl-enzyme, and the phosphorolysis step, and if the initial binding of 3-PGA occursat the Pi site, the subsequent “flipping” takes place at the stage preceding hydride ion transfer.
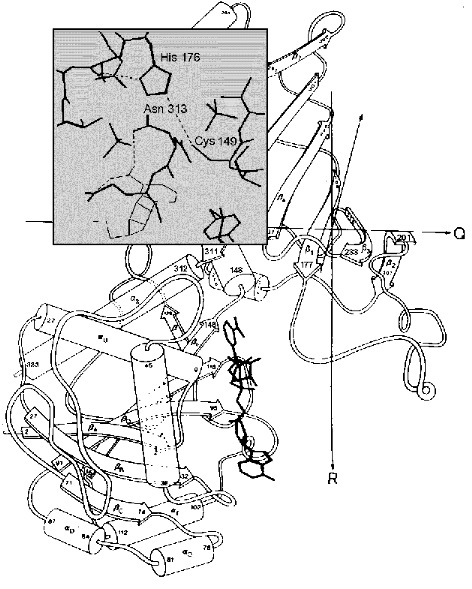
This would be in line with the hypothetical mechanism suggested recentlyby the authors of the above-mentioned crystallographic investigation of the complexes formed by E. coli GAPDH with the substrate and the coenzyme [31]. A superposition of the binary enzyme*NAD+ complex structure on the structure of the apoenzyme-thiohemiacetal complex (where thiohemiacetal was bound at the Pi site) revealed an interaction between an oxygen in the hydroxy group at the first carbon atom of the thiohemiacetal and the nicotinamide ring of NAD+. This points to the likelihood of NAD+ participating in the stabilization of the transition state of the thiohemiacetal formation reaction that proceeds at the Pi site and is in agreement with the idea that an initial binding of substrate occurs at this site. It is to be noted, however, that the model discussed (shown schematically in Fig. 3b) assumes that the moving of the substrate phosphate group to the Ps site precedes the hydride ion transfer step, i.e., the stage of acyl-enzyme*NADH complex formation. This becomes possible probably due to local conformational changes in the flexible loop connecting the beta2-strand and the 212-214 helix (see Fig. 4), which brings the cluster of residues comprising the Pi site closer to the catalytically important Cys149 [31].
Therefore, one of the reasons why initial substrate binding occurs at the Pi site may be to create an environment most favorable for the formation of thiohemiacetal. Yet subsequent conversions of this reaction intermediate take place only after the binding of the substrate phosphate group at the Ps site; during the “flipping” process (Fig. 3b, step 2) the conformation of the thiohemiacetal molecule changes through rotation around the bond between the C1 and C2 atoms [31]. As noted above, this hypothetical scheme, based on structural studies of E. coli GAPDH, fits in with the results of our research on another mesophilic enzyme, rabbit muscle GAPDH, which has shown that chemical modification of Arg231 located at the Ps site considerably slows down the oxidative step of the reaction (step 3 in Fig. 3b).Fig. 4. A part of the GAPDH tertiary structure. Amino acid residues 1-147 and 312-333 form the NAD+-binding domain and residues 148-311, the catalytic domain. Bold lines show the structure of NAD+. Taken from [43] with alterations. Insert: the active site region including Cys149 and His176, and also Asn313, which holds the carboxyamide group of the NAD+ molecule in the correct position. Hydrogen bonds are shown by dotted lines. Taken from [2] with alterations.
However, an alternative version of that scheme (see Fig. 3a) appeared to better explain the results of kinetic experiments with B. stearothermophilus GAPDH [30]. In the absence of structural data on the ternary complexes formed at different steps of the reaction, it is difficult to find a satisfactory reason for these discrepancies. At the same time, the amount of information amassed to date which points to a “flipping” of the substrate phosphate group from one anion-binding site to another leaves little doubt that this process could bean important component of the catalytic mechanism possibly involved in the regulation of the enzyme's functioning.
TWO MECHANISMS FORMING THE BASIS OF THE FUNCTIONAL
NON-EQUIVALENCE OF GAPDH ACTIVE CENTERS: COOPERATIVITY OF
NAD+ BINDING AND HALF-OF-THE-SITES REACTIVITY
One important problem, long discussed in literature, has to do with a possible interrelation between the mechanisms that account for different manifestations of nonequivalence of the active centers within a tetrameric GAPDH molecule. According to the concept proposed by Levitzki et al. [34-36], both the negative cooperativity in the NAD+ binding and the half-of-the-sites reactivity result from conformational changes induced by the interaction of a ligand (NAD+ or a “half-of-the-sites reagent”) with the adenosine-phosphate binding region of the active center and transmitted to neighboring subunits. An alternative model developed in the studies of Bernhard et al. [4, 6, 9, 14] is based on the assumption that half-of-the-sites reactivity is the result of a preexisting asymmetry of the tetrameric GAPDH molecule, which is built as a dimer of dimers; this structural feature may also explain negative cooperativity in the binding of coenzyme.
Studies performed in our laboratory have made some contribution to the understanding of the structural basis of these phenomena. This applies in particular to elucidating the character of apoenzyme-coenzyme interaction. A combination of experimental approaches developed in the laboratory, including an array of different spectral methods and the use of anionic and cationic fluorescent dyes, as well as various fragments of the NAD+ molecule, made it possible to follow conformational changes induced by the binding of specific ligands [37, 38].
Structural alterations of the adenosine-binding site of the active center induced by the binding of the nicotinamide fragment of the NAD+ molecule were revealed [39, 40]. The results obtained in these studies were the first demonstration of the role of the nicotinamide component of the coenzyme molecule in defining the conformational transitions accompanying the GAPDH-NAD+ interaction and in generating cooperative effects. Several years later, Henis and Levitzki [41] and other authors [42] confirmed our conclusion and presented further evidence supporting the notion that cooperativity in NAD+ binding results from successive conformational changes of the oligomeric enzyme molecule induced by the binding of the ligand and determined by the interaction of the protein with the nicotinamide part of the coenzyme.
Results of X-ray crystallography analysis have made it possible to describe in detail the interaction of this part of the NAD+ molecule with the GAPDH active center, revealing the critical role of the hydrogen bond formed between the carboxyamide group of the nicotinamide ring and Asn313 [1, 3]. Figure 4 shows the structure of B. stearothermophilus GAPDH monomer; in the insert (covering a part of the catalytic domain) one can see the active center region including Cys149 and His176, whose correct mutual orientation is essential for catalysis, as well as the nicotinamide ring of NAD+ (depicted by thin lines in the lower part of the insert). The dotted lines represent hydrogen bonds. Along with the hydrogen bond between Cys149 and His176, the hydrogen bonds that stabilize the carboxyamide group in its correct position are also shown (one of these bonds was mentioned above).
Using differential scanning calorimetry, we tried to obtain additional information on the involvement of the nicotinamide portion of NAD+ in the coenzyme's effect on interdomain interactions within a monomer. It was shown [44] that the binding of NAD+ considerably affects the thermal denaturation parameters of the enzyme (the maximal temperature of thermal unfolding and the calorimetric enthalpy are increased). Another manifestation of the NAD+ effect was a marked increase in the cooperativity of the conformational transition, which might point to a coenzyme-induced narrowing of the gap between the catalytic and the NAD+-binding domains, thus providing experimental evidence of the phenomenon described on the basis of crystallographic studies [2].
To elucidate the role of the nicotinamide ring of NAD+ in the conformational changes, we applied an indirect approach, chemically modifying Cys149 in the immediate vicinity of Asn313, i.e., of the residue that ensures the correct positioning of the nicotinamide ring by forming a hydrogen bond with its carboxyamide group (see Fig. 4, insert). Replacement of Cys149 by Ser (in B. stearothermophilus GAPDH) caused virtually no change in the thermal unfolding parameters of the apoenzyme and in the effect of NAD+. At the same time, carboxymethylation of Cys149, which did not prevent the enzyme from binding NAD+, destabilized the protein and led to the disappearance of the coenzyme's effect on the cooperativity of the thermal unfolding transition [44]. It seems likely that the above alterations could have resulted from the steric effect of the bulky carboxymethyl group, which hindered the correct positioning of the nicotinamide ring, blocking its influence on the protein conformation. Given the character of interdomain interactions in the tetrameric GAPDH molecule (see Fig. 1), one might expect that changes in such interactions within separate monomers would influence, to a greater or lesser extent, also the intersubunit domain-domain interactions. On this assumption, the results of our study can be interpreted as supporting the idea of the nicotinamide ring playing a decisive role in the cooperativity of NAD+ binding by the tetramer.
Elucidation of molecular mechanisms underlying the cooperativity of GAPDH active centers in the binding of NAD+ remains a problem to be investigated. One approach to gathering information on this subject is to use differential scanning calorimetry in combination with mutagenesis of amino acid residues involved in intramolecular interactions of different types, with the aim of differentiating between the effect induced by the coenzyme within a single monomer and within a tetrameric enzyme molecule, and to obtain evidence for transmission of ligand-induced conformational changes via subunit contacts of different types. Working in this direction, we have obtained results which point to a significant role of the intersubunit interactions formed between the catalytic domains of adjacent monomers (i.e., between O and P, and Q and R subunits respectively, see Fig. 1) in the stabilization of the tetramer in its holo-conformation.
These preliminary results may be helpful in interpreting the observation that mutation of His176 located in the catalytic domain drastically alters the effect of NAD+ on the conformational state of the tetramer. Namely, not only does the binding of NAD+ to a His176Asn mutant fail to cause a narrowing of the thermal transition peak (suggesting an increase in the cooperativity of thermal unfolding), but on the contrary, it markedly increases the width of the peak [44]. Apparently the mutation, while lowering the cooperativity of the catalytic domain's transition into the holo-state, which is induced by the binding of NAD+ at the nucleotide-binding domain of the same subunit, also disrupts the transmission of conformational changes between adjacent subunits via their catalytic domains. The results of this investigation are consistent with the model in which the successive conformational changes induced by the binding of NAD+ to the tetrameric GAPDH molecule are the structural basis for the nonequivalence of its active centers.
Let us now turn to another type of functional nonequivalence of GAPDH active centers, which is half-of-the-sites reactivity. What is special about this property of the enzyme is the fact that it manifests itself upon interaction with certain substrate analogs and inhibitors which bind in the active centers, but not with the natural substrate, 3-PGA [7, 9, 13]. This complicates the interpretation of the functional role of the active centers' nonequivalence; at the same time, the ability of the tetramer to become stabilized in an asymmetric state attracts interest to thestructural basis and possible functional significance of such stabilization. As pointed out before, one of the models explaining the origin of half-of-the-sites reactivity postulates a preexistingasymmetry of the tetramer and the existence of an equilibrium between its symmetric and asymmetric states [6].
This model is supported by a large body of experimental data [4, 6, 9, 11, 14, 45] that, however, contain no information on the structural basis of such a phenomenon, i.e., on the amino acid residues involved in the assumed transition of the tetramer from a symmetric to an asymmetric state. We have been able to demonstrate that one of those residues is Arg231, whose role in the active center is discussed above. It has been shown that incubation of rabbit muscle GAPDH in the presence of 2,3-butanedione, resulting in the modification of one arginine residue per subunit of the tetramer (as noted before, only Arg231 becomes modified under the conditions used) not only caused a significant drop in catalytic activity, but also stabilized the tetramer in an asymmetric state [28, 46-48]. Two active centers of the modified enzyme retained the ability to catalyze the reaction of 3-PGA oxidation (with activity lowered by the arginine modification), while the other two active centers appeared nonfunctional. In these experiments, half-of-the-sites reactivity was demonstrated towards the natural substrate.
The modified enzyme exhibited half-of-the-sites reactivity also towards a number of reagents interacting with functionally important Cys149. Thus, data obtained upon titration of the modified enzyme with 5,5´-dithionitrobenzoate (DTNB) indicated that the SH-groups of two Cys149 residues per tetramer had been inaccessible to modification. A similar situation was observed in experiments with alkylating agents selectively modifying Cys149, namely iodoacetate and iodoacetamide.
Aside from the physiological reaction (oxidative phosphorylation of 3-PGA), we also examined GAPDH-catalyzed hydrolysis of p-nitrophenyl acetate, which proceeds in the absence of NAD+ and includes two steps: acylation of Cys149 and subsequent hydrolysis of the acetyl-enzyme. It has been found that chemical modification of Arg231 has no effect on the rate of that reaction, which was to be expected given the functional role of this residue; however, the number of simultaneously working active centers is reduced by half [46]. This suggests that modification of Arg231, both in this case and in the experiments with GAPDH catalyzing the physiological reaction, imposed some conformational restrictions that prevented half of the active centers from functioning. As to the catalytic efficiency of the other half of the active centers, it depends on the role played by Arg231 in the reaction observed; in the case of 3-PGA oxidation, the catalytic rate is 5-7% of the maximum, while in the case of p-nitrophenyl acetate hydrolysis it approaches 100%.
Similar results have been obtained with GAPDH from another source, E. coli [49-51]. In this case the active centers unable to perform the oxidative reaction appeared accessible to alkylating agents (iodoacetate and iodoacetamide). Hence, one may conclude that (a) the SH-groups of the active centers incapable of functioning in the oxidative reaction remain intact, and (b) some minor conformational differences should exist between the active centers of rabbit muscle and E. coli GAPDHs, which make them unequally accessible to alkylating agents upon modification of Arg231.
A study into the coenzyme-binding properties of the enzyme has shown that these properties are practically unchanged by the modification of arginine residues: both the stoichiometry of the binding (4 NAD+ equivalents per mol) and the negative cooperativity (first two NAD+ equivalents bound with a Kd of about 0.01 µM, and the third and fourth ones with a Kd of about 1-3 µM [46]). These properties of the enzyme could have one of the following two explanations: 1) modification of the arginine residues stabilizes the tetramer in a new conformational state, with highly expressed negative cooperativity of active centers within each dimer. Consequently, a catalytic conversion in one of those active centers precludes the functioning of the adjacent one; the active centers of such a dimer exhibit negative cooperativity also in the binding of NAD+; 2) the tetramer is characterized by preexistent asymmetry and exists in both the symmetric and the asymmetric states, which are in equilibrium. Arg231 is involved in the conformational transition between the two states, and its modification locks the tetramer in the asymmetric state. Transition from the symmetric to the asymmetric state is not connected with alterations of the coenzyme-binding properties of the tetramer.
To rule out one of these possibilities, complexes of the native and modified apo-GAPDHs with NAD+, containing different amounts of bound coenzyme, were prepared and tested for their ability to form an acyl-enzyme*NADH complex in the presence of an excess of substrate. Figure 5 presents the results obtained. It is seen that with two NAD+ equivalents added per mol tetramer and with an excess of substrate, reaction took place in two active centers of the native enzyme, but only in one active center of the modified enzyme. The fact that the coenzyme-binding characteristics were not changed upon modification means that two NAD+ equivalents were bound per tetramer in both cases. Because the catalytic reaction took place in only one of the centers occupied by the coenzyme, it appears that the distribution of NAD+ between the active centers of the modified enzyme was random, i.e., did not depend on the ability of the centers to catalyze the oxidative reaction. This is schematically illustrated in the right-hand part of the figure.
The results of this investigation support the idea that the mechanisms that give rise to negative cooperativity in the binding of NAD+ and those behind half-of-the-sites reactivity are not the same. In the former case, as discussed above, successive conformational changes are induced by the binding of NAD+ to individual subunits of the tetramer, whereas in the latter case, half-of-the-sites reactivity probably arises from the preexisting asymmetry of the tetramer. The experimental approaches used in our study allowed stabilization of the enzyme in its asymmetric state and identification of the amino acid residue involved in the conformational transition between the symmetric and asymmetric states, thus substantiating the model proposed by Bernhard and Seydoux [6, 52].Fig. 5. Reaction of apoenzyme*NAD+ complexes containing different amounts of bound NAD+, with D-glyceraldehyde-3-phosphate. 1) Native enzyme; 2) modified enzyme. The samples containing one or two NAD+ equivalents per tetramer were prepared by adding corresponding amounts of the coenzyme to native or modified apo-GAPDH solutions. The samples containing three or four NAD+ equivalents per tetramer were prepared using various holoenzyme preparations from rabbit muscle. The formation of the apoenzyme*NAD+ complex was followed by absorbance at 360 nm; that of the acyl-enzyme*NADH complex was followed by absorbance at 340 nm. The concentration of GAPDH was 10 µM [47]. Shown schematically in the right-hand part of the figure are possible distributions of NAD+ between the subunits of a modified enzyme containing two equivalents of bound coenzyme per tetramer. The conformation of active subunits is represented by circles, and that of inactive ones, by squares. The reaction can only take place if NAD+ is bound in a “circular-shaped” subunit. Figures to the right of the tetramer diagrams indicate the number of functioning active centers.
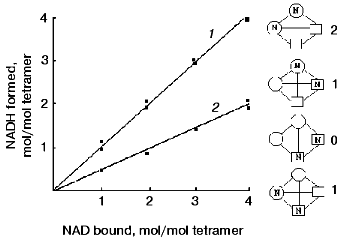
Assuming that chemical modification of Arg231 stabilizes the asymmetric state, we suggested that the transition between the symmetric and asymmetric states should be accompanied by certain alterations in the neighborhood of this residue [46]; however, in the absence of crystallographic data on the objects of our investigation (rabbit muscle and E. coli GAPDHs), no definite conclusion could be made. Several years after the completion of this work, an X-ray crystallography study was performed on E. coli GAPDH [3], yielding results that appeared to be in a good agreement with our concept. Specifically, a preexistent asymmetry of the tetramer was revealed, which manifested itself in a minor structural difference between the O and Q subunits on one hand, and the P an R subunits on the other hand (see Fig. 1). The difference resides in the interaction of Arg231 located within the beta3 strand (see Fig. 4) with amino acid residues of the so-called “S-loop”, which connects beta1 and beta2. While in the P and R subunits Arg231 is hydrogen-bonded only with Thr179, two alternative possibilities exist in the O and Q subunits, where Arg231 may establish a hydrogen bond either with Thr179 or with Asp192. In the first case, all subunits become conformationally identical, and a symmetric tetramer is formed; in the second case, the asymmetric structure is stabilized. These data give reasons to suggest that chemical modification of Arg231 “locks” the tetramer in one of its two alternative states (the asymmetric one), precluding a transition to the other state.
In contrast to the enzyme isolated from E. coli, GAPDH from B. stearothermophilus exhibits no preexisting asymmetry [1, 3]. One possible explanation is that amino acid sequences in the region of the S-loop are markedly different in these two proteins [53]. In B. stearothermophilus GAPDH, the very nature of the amino acid sequence around residues 187-191 precludes any hydrogen bond between Arg231 and Asp192 in any of the subunits of a tetramer; Arg231 can only be bound to Thr179 [1, 3]. It is interesting to note that no half-of-the-sites reactivity is observed with this enzyme under the conditions where rabbit muscle GAPDH exhibits this phenomenon [54].
Despite extensive studies on half-of-the-sites reactivity, not much can be said about the physiological significance of this property. In the case of GAPDH, no half-of-the-sites reactivity has been observed in kinetic experiments with 3-PGA, although some experimental data point to a change in the number of simultaneously functioning active centers in the native GAPDH molecule upon transition from the pre-steady state to the steady state phase of reductive dephosphorylation of 1,3-bisphosphoglycerate [11]. One hypothetical reason may be that an asymmetric tetramer is a transient intermediate in the conformational transitions accompanying certain steps of catalysis.
In conclusion, it is worth noting that half-of-the-sites reactivity, a well-known property of many dimeric and poly-dimeric enzymes, for the most part remains poorly understood in its structural aspect. Some progress has been achieved in a few cases where half-of-the-sites reactivity was shown to result from an inherent pair-wise asymmetry of the enzyme molecule [51]. A legitimate question arises as to the role this phenomenon may play in the regulation of holo-oligomer activity(by inducing reversible changes in the number of simultaneously functioning active centers) and as to the mechanisms through which this regulation is accomplished.
The experimental approach employed in our study to reveal the preexistent asymmetry of GAPDH is an artificial ploy, although given the abundance of arginine-modifying enzymes actually present in the cells [55-57], one could imagine a theoretical possibility that post-translational modification of an enzyme at arginine residues might serve as a mechanism for its regulation. In the case of GAPDH, however, such a mechanism would hardly be effective, since along with changing the number of functioning active centers it would also cause a considerable degree of inactivation in those centers still able to function.
It seems plausible that other mechanisms may exist for regulating the number of simultaneously functioning active centers. In particular, this possibility is being considered in connection with the molecular mechanisms underlying the half-of-the-sites reactivity of mitochondrial aldehyde dehydrogenase. This tetramer is built as a dimer of dimers, exhibits no active center cooperativity in catalysis, yet clearly displays half-of-the sites reactivity [58]. Recently, with the use of mutant and hybrid enzyme forms, it became possible to elucidate the mechanism switching theenzyme from the “half-of-the-sites” to the “all-of-the-sites” functioning mode [59].
All of the above deals with just one area of studies by a research team founded by Professor Severin. Beside the author of this review, it included Dr. R. A. Asryants, Dr. M. V. Ivanov, Dr. V. I. Muranetz, Dr. L. I. Ashmarina, and Dr. E. V. Kuzminskaya (Schmalhausen), as well as undergraduate and postgraduate students and laboratory assistants. Their respective contributions to the work reviewed here are reflected in the corresponding literature (see references below). Regrettably, volume restrictions have made it impossible to cover the results obtained in other research areas (such as the role of protein-protein interactions of various complexity in the functioning of D-glyceraldehyde-3-phosphate dehydrogenase), which were equally of interest to Professor Severin and are being actively developed in the laboratory.
Funding for the work was provided by the State Program for the Maintenance of Scientific Schools (grant No. 00-15-97758) and the Russian Foundation for Basic Research (grant No. 99-04-48076).
REFERENCES
1.Skarzynski, T., Moody, P. C., and Wonacott, A. J.
(1987) J. Mol. Biol., 193, 171-187.
2.Skarzynski, T., and Wonacott, A. J. (1988) J.
Mol. Biol., 203, 1097-1118.
3.Duee, E., Olivier-Deyris, L., Fanchon, E., Corbier,
C., Branlant, G., and Dideberg, O. (1996) J. Mol. Biol.,
257, 814-838.
4.Bernhard, S. A., and MacQuarrie, R. A. (1973) J.
Mol. Biol., 74, 73-78.
5.Koshland, D. E., Jr., Nemethy, G., and Filmer, D.
(1966) Biochemistry, 5, 365-385.
6.Seydoux, F., Malhotra, O. P., and Bernhard, S. A.
(1974) in CRC Critical Reviews in Biochemistry (Fasman, G. D.,
ed.) Vol. 2, CRC Press, Cleveland, Ohio, pp. 227-257.
7.Stallcup, W. B., and Koshland, D. E., Jr. (1973)
J. Mol. Biol., 80, 41-62.
8.Convay, A., and Koshland, D. E., Jr. (1968)
Biochemistry, 7, 4011-4023.
9.Malhotra, O. P., and Bernhard, S. A. (1973)
Proc. Natl. Acad. Sci. USA, 70, 2077-2081.
10.Cardon, J. W., and Boyer, P. D. (1982) J.
Biol. Chem., 257, 7615-7622.
11.Kellershohn, N., and Seydoux, F. J. (1979)
Biochemistry, 18, 2465-2470.
12.Stallcup, W. B., and Koshland, D. E., Jr. (1972)
Biochem. Biophys. Res. Commun., 49, 1108-1114.
13.Byers, L. D., and Koshland, D. E., Jr. (1975)
Biochemistry, 14, 3661-3669.
14.Malhotra, O. P., Bernhard, S. A., and Seydoux, F.
(1981) Biochimie, 63, 131-141.
15.Levitzki, A., and Koshland, D. E., Jr. (1978)
Curr. Topics Cell Regul., 10, 1-40.
16.Buehner, M., Ford, G. C., Moras, D., Olsen, K.
W., and Rossmann, M. G. (1974) J. Mol. Biol., 90,
25-49.
17.Segal, H. L., and Boyer, P. D. (1953) J. Biol.
Chem., 204, 265-281.
18.Garavito, R. M., Rossmann, M. G., Argos, P., and
Eventoff, W. (1977) Biochemistry, 16, 5065-5071.
19.Branden, C.-I., and Eklund, H. (1980) in
Dehydrogenases Requiring Nicotinamide Coenzymes (Jeffery, J.,
ed.) Birkhauser Verlag, Basel, pp. 41-84.
20.Nagradova, N. K., and Asryants, R. A. (1975)
Biochim. Biophys. Acta, 386, 365-368.
21.Nagradova, N. K., Asryants, R. A., Benkevich, N.
V., and Safronova, M. I. (1976) FEBS Lett., 69,
246-248.
22.Nagradova, N. K., Asryants, R. A., and Benkevich,
N. V. (1978) Biochim. Biophys. Acta, 527, 319-326.
23.Asryants, R. A., Benkevich, N. V., and Nagradova,
N. K. (1983) Biokhimiya, 48, 193-200.
24.Asryants, R. A., Rychkova, O. Yu., and Nagradova,
N. K. (1983) Biokhimiya, 48, 531-538.
25.Douzhenkova, I. V., Asryants, R. A., Muronetz, V.
I., and Nagradova, N. K. (1986) Biokhimiya, 51,
1899-1907.
26.Douzhenkova, I. V., Asryants, R. A., and
Nagradova, N. K. (1988) Biochim. Biophys. Acta, 957,
60-70.
27.Asryants, R. A., Ashmarina, L. I., Muronetz, V.
I., and Nagradova, N. K. (1980) FEBS Lett., 118,
141-144.
28.Kuzminskaya, E. V., Asryants, R. A., and
Nagradova, N. K. (1991) Biochim. Biophys. Acta, 1075,
123-130.
29.Banas, T., Krotkiewska, B., Marcinkowska, A., and
Wolny, M. (1983) Acta Biochem. Pol., 30, 324-334.
30.Corbier, C., Michels, S., Wonacott, A. J., and
Branlant, G. (1994) Biochemistry, 33, 3260-3265.
31.Yun, M., Park, C.-G., Kim, J.-Y., and Park, H.-W.
(2000) Biochemistry, 39, 10702-10710.
32.Nagradova, N. K., and Schmalhausen, E. V. (1998)
Biochemistry (Moscow), 63, 504-515.
33.Asryants, R. A., Kuzminskaya, E. V., Tishkov, V.
I., Douzhenkova, I. V., and Nagradova, N. K. (1989) Biochim.
Biophys. Acta, 997, 159-166.
34.Levitzki, A. (1973) Biochem. Biophys. Res.
Commun., 54, 889-893.
35.Schlessinger, J., and Levitzki, A. (1974) J.
Mol. Biol., 82, 547-561.
36.Levitzki, A. (1974) J. Mol. Biol.,
90, 451-458.
37.Ivanov, M. V., Klichko, V. I., Nikulin, I. R.,
Asryants, R. A., and Nagradova, N. K. (1982) Eur. J. Biochem.,
125, 291-297.
38.Klichko, V. I., Ivanov, M. V., and Nagradova, N.
K. (1986) Biokhimiya, 51, 465-475.
39.Ivanov, M. V., Asryants, R. A., and Nagradova, N.
K. (1976) Int. J. Biochem., 7, 473-478.
40.Ivanov, M. V., and Nagradova, N. K. (1977)
Biokhimiya, 42, 211-222.
41.Henis, Y. I., and Levitzki, A. (1980) Eur. J.
Biochem., 112, 59-73.
42.Gloggler, K. G., Balasubramanian, K., Beth, A.,
Park, J. H., and Trommer, W. E. (1982) Biochim. Biophys. Acta,
706, 197-202.
43.Biesecker, G., Harris, J. I., Tierry, J. C.,
Walker, J. E., and Wonacott, A. J. (1977) Nature, 266,
328-333.
44.Levashov, P. A., Orlov, V. N., Boschi-Muller, S.,
Talfournier, F., Asryants, R. A., Bulatnikov, I. G., Muronetz, V. I.,
Branlant, G., and Nagradova, N. K. (1999) Biochim. Biophys.
Acta, 1433, 294-306.
45.Ho, Y.-S., and Tsou, C.-L. (1979) Nature
(London), 277, 245-246.
46.Kuzminskaya, E. V., Asryants, R. A., and
Nagradova, N. K. (1992) Biochem. Biophys. Res. Commun.,
187, 577-583.
47.Nagradova, N. K., Kuzminskaya, E. V., and
Asryants, R. A. (1993) Biotechnol. Appl. Biochem., 18,
157-163.
48.Nagradova, N. K., Asryants, R. A., Kuzminskaya,
E. V., Ashmarina, L. I., and Muronetz, V. I. (1996) in Chemical
Modification of Enzymes (Kurganov, B. I., Nagradova, N. K., and
Lavrik, O. I., eds.) Nova Science Publishers, Inc., New York, pp.
59-125.
49.Levashov, P. A., Schmalhausen, E. V., Muronetz,
V. I., and Nagradova, N. K. (1995) Biochem. Mol. Biol. Int.,
37, 991-1000.
50.Nagradova, N. K., Schmalhausen, E. V., Levashov,
P. A., Asryants, R. A., and Muronetz, V. I. (1996) Biotechnol. Appl.
Biochem., 61, 47-56.
51.Nagradova, N. K. (2000) FEBS Lett.,
487, 327-332.
52.Viratelle, O. M., and Seydoux, F. (1975) J.
Mol. Biol., 92, 193-205.
53.Martin, W., and Cerff, R. (1986) Eur. J.
Biochem., 159, 323-331.
54.Ho, Y.-S., Liang, S. J., and Tsou, C. L. (1980)
Biochim. Biophys. Acta, 613, 249-255.
55.Aletta, J. M., Cimato, T. R., and Ettinger, M. J.
(1998) Trends Biochem. Sci., 23, 89-91.
56.Tang, J., Kao, P., and Herschman, H. R. (2000)
J. Biol. Chem., 275, 19866-19876.
57.Zhang, X., Zhou, L., and Cheng, X. (2000) EMBO
J., 19, 3509-3519.
58.Dryjanski, M., Lehmann, T., Abriola, D., and
Pietruszko, R. (1999) J. Prot. Chem., 18, 627-636.
59.Zhou, J., and Weiner, H. (2000)
Biochemistry, 39, 12019-12024.