Macroscopic Aggregation of Tobacco Mosaic Virus Coat Protein
V. N. Orlov1, A. M. Arutyunyan1, S. V. Kust1, E. A. Litmanovich2, V. A. Drachev1, and E. N. Dobrov1*
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: dobrov@pcman.genebee.msu.su2Department of Polymer Sciences, School of Chemistry, Lomonosov Moscow State University, Moscow, 119899 Russia
* To whom correspondence should be addressed.
Received October 5, 2000; Revision received November 5, 2000
The relationship between processes of thermal denaturation and heat-induced aggregation of tobacco mosaic virus (TMV) coat protein (CP) was studied. Judging from differential scanning calorimetry “melting” curves, TMV CP in the form of a trimer-pentamer mixture (“4S-protein”) has very low thermal stability, with a transition temperature at about 40°C. Thermally denatured TMV CP displayed high propensity for large (macroscopic) aggregate formation. TMV CP macroscopic aggregation was strongly dependent on the protein concentration and solution ionic strength. By varying phosphate buffer molarity, it was possible to merge or to separate the denaturation and aggregation processes. Using far-UV CD spectroscopy, it was found that on thermal denaturation TMV CP subunits are converted into an intermediate that retains about half of its initial alpha-helix content and possesses high heat stability. We suppose that this stable thermal denaturation intermediate is directly responsible for the formation of TMV CP macroscopic aggregates.
KEY WORDS: tobacco mosaic virus coat protein, thermal denaturation, partially folded intermediate, aggregation, circular dichroism, differential scanning calorimetry
Abbreviations: TMV) tobacco mosaic virus; CP) coat protein; HP) helical protein; PUF) partially unfolded form; PB) phosphate buffer; DSC) differential scanning calorimetry.
Formation of large “macroscopic” aggregates has been often
observed in the course of protein denaturation and has always been
considered as a nuisance and a source of various artifacts. But in
recent years this process has started to attract much attention [1, 2] because of its probable role
in prion infections [3] and in so-called
“amyloid” disease progression [4, 5], as well as in formation of inclusion bodies during
the expression of recombinant proteins in bacteria [6].
For many years, coat protein (CP) of tobacco mosaic virus (TMV) has served as a model in different kinds of structural studies. Depending on conditions (pH, temperature, ionic strength, protein concentration), TMV CP forms in solution several types of specific ordered aggregates [7, 8]. In particular, at pH >= 8.0 and low ionic strength TMV CP exists in the form of dynamic mixture of pentamers and trimers with a small content of monomers, this being called A- or 4S-protein. At pH near 7.0 and ionic strength of about 0.1 M, TMV CP is transformed into the 20S-aggregate form (two-layer disk or short helix) with an admixture of 4S-protein, and at pH <= 6.0 the protein produces long virus-like helical aggregates called repolymerized protein [7, 8]. But besides this ordered aggregation (polymerization), TMV CP has turned out to be highly proficient in macroscopic (large “unspecific”) aggregate formation [9, 10].
Recently, Fink [1] hypothesized that, in contrast to previous ideas, macroscopic aggregate formation results from specific intermolecular interactions of hydrophobic surfaces of partly folded protein molecule subdomains that are normally involved in intramolecular interactions.
In the present work, we studied TMV CP thermal denaturation, thermally induced macroscopic aggregation, and the relationship between these two processes. In this study, a stable partially unfolded intermediate was identified that may directly participate in the formation of TMV CP macroscopic aggregates.
MATERIALS AND METHODS
TMV purification and TMV CP preparation. The wild-type (strain U1) TMV was propagated in Nicotiana tabacum var. Samsun plants and purified by standard methods [10]. TMV CP was prepared by the acetic acid method [11] and stored at concentration of about 5 mg/ml in 5 mM phosphate buffer (PB) pH 8.0 at 4°C. The protein concentration was measured by UV spectroscopy using absorption coefficient E0.1%280 = 1.30 [12].
UV spectroscopy. Absorption spectra, time dependence of macroscopic aggregation at constant temperature, and heating curves were measured in 0.5- or 1-cm cells in a Specord UV-VIS (Carl Zeiss, Germany) spectrophotometer. All aggregation kinetics measurements were performed at 52°C. PB samples were preheated for 10 min, and then aliquots (13 to 80 µl) preheated at 35°C of a stock TMV CP solution were added to the final volume of 1 ml (0.5-cm cells) or 2 ml (1-cm cells), thoroughly mixed (dead time 20 sec), and the time-course of “absorption” increase at 313 nm was monitored. The initial rate of aggregation (Vin) was calculated in optical units per minute from the initial linear part of the curve.
In melting experiments, TMV CP samples (200 µg/ml) in 1-cm cells were heated for 20 min in 3 to 5°C steps in temperature-controlled cells of the Specord UV-VIS. The aggregation curves display the temperature dependence of “absorption” (turbidity) at 313 nm. The temperature in the cell was measured with a copper-constantan thermocouple.
Differential scanning calorimetry. Calorimetric measurements were carried out in a DASM-4 differential adiabatic scanning calorimeter (Biopribor, Russian Academy of Sciences, Pushchino, Russia) with a 0.47-ml capillary platinum cell interfaced with an IBM-compatible computer. The heating rate was 0.125 to 1°C/min. The reversibility of the thermal transitions was checked by reheating the samples after cooling from the first scan. Because the thermal transition of TMV CP was completely irreversible, the calorimetric traces were corrected for the instrumental baseline by subtracting scans for the reheating of the samples. A constant pressure of 2 atm was always maintained to prevent possible degassing of the samples on heating. TMV CP molecular weight was taken to be 17.5 kD.
CD spectroscopy. CD spectra in the 198 to 250 nm region were measured in a modified Jobin-Ivon Mark V (France) dichrograph interfaced with an IBM-compatible PC using the RDA and Wtest programs developed in our laboratory. Measurements were performed in 1-mm cells at TMV CP concentration 200 µg/ml.
Analytical centrifugation. Analytical centrifugation of TMV CP samples (33 to 200 µg/ml) was performed in a Beckman E (USA) ultracentrifuge equipped with a scanner at 280 to 350 nm, 20°C, and 2,000 to 36,000 rpm.
Dynamic light scattering. Dynamic light scattering of TMV CP aggregates was measured with an ALV-5 photogoniodiffusometer (ALV, Germany) using a 25 mW 633 nm He-Ne laser at five concentrations in the 12- to 100-µg/ml range. Correlation functions were calculated using a 72-channel Photocor-M correlometer, and hydrodynamic radius (Rh) of the aggregates was determined by the cumulants method and by Tikhonov regularization [13].
RESULTS
1. Thermal denaturation of tobacco mosaic virus coat protein. Most of the experiments described below were performed at pH 7.0 or 8.0. PB (50 mM, pH 7.0) and temperature of 20°C can be considered as “physiological conditions” for TMV CP because under these conditions the protein most efficiently forms virions in vitro with homologous RNA [7, 8]. But under these conditions, TMV CP exists in solution as a mixture of 4S and 20S aggregates, with each aggregate fraction strongly depending on the protein concentration, temperature, and other factors [7, 8]. At pH 8.0, TMV CP exists as “pure” 4S aggregates, which facilitates interpretation of the results.
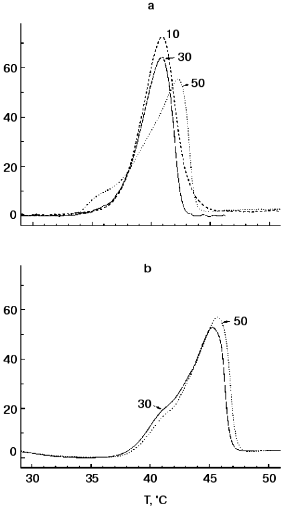
Figure 1 shows DSC “melting curves” of TMV CP at pH 7.0 and 8.0 under standard conditions (concentration, 2 mg/ml; heating rate, 1°C/min). It can be seen that in 50 mM PB, pH 8.0, TMV CP undergoes highly cooperative irreversible melting at temperatures as low as 42.0°C with DeltaH of 250 kcal/mol and DeltaTmax of 4°C (Fig. 1a). At pH 7.0, the heat absorption curve had two components with a minor peak at 42.0°C and major one at 45.5°C (Fig. 1b). In all probability, the major peak (80% of the total heat absorption) corresponds to melting of CP subunits in 20S aggregates, and the minor peak (20%) to melting of CP molecules in an admixture of 4S aggregates. Thus, 20S aggregates posses only small additional stability compared to 4S aggregates and have Tmax much lower than that of TMV CP virus-like helical polymer (HP) (73°C [14, 15]). On decreasing the ionic strength to 30 or 10 mM, at pH 8.0, the TMV CP melting temperature decreased slightly (by 1°C) (see also [14, 15]), and at pH 7.0 in 30 mM buffer no changes in the DSC scan compared to 50 mM PB were observed (Fig. 1b). No additional endothermic or exothermic peaks were found on further heating of TMV CP samples at pH 7.0 or 8.0 up to 100°C. DSC melting of TMV CP is irreversible under all heating regimes and concentrations tested [14, 15].
Figure 2 presents TMV CP (2 mg/ml) melting curves in 30 mM PB, pH 8.0, at different heating rates and protein concentrations. As is often observed for kinetically controlled melting [16, 17], decrease in the heating rate from 1 to 0.125 °C/min resulted in a decrease in the melting temperature from 40.9 to 37.7°C (Fig. 2a). At the same time, TMV CP displayed only a small (about 0.4°C) decrease in melting temperature on decrease in concentration from 2.0 to 0.5 mg/ml (Fig. 2b).Fig. 1. TMV CP heat absorption curves in PB, pH 8.0 (a) and 7.0 (b). Figures on the curves correspond to PB concentrations (in mM). Scanning rate, 1°C/min; protein concentration, 2 mg/ml.
The thermal stability of TMV CP subunits in small (4S and 20S) aggregates observed in DSC experiments seems to be surprisingly low, but it agrees well with the results of the test on functional activity of TMV CP developed by Jockusch about 40 years ago [18, 19]. In this test, TMV samples (in different buffer solutions) are heated to increasing temperatures in 20-min 2-3°C steps, and their ability to form virus-like HP after adjusting the pH to 5.1 is checked by low-speed (1500g) centrifugation. Normal HP has a sedimentation coefficient of about 150S and is pelleted only at about 100,000g. But if the CP was denatured by prior heating, it produces at pH 5.1 large nonspecific aggregates which completely sediment at 1500g. As shown in Jockusch's works [18, 19] and in our experiments [9, 10], for TMV CP at pH 7 to 8 and low or moderate ionic strength, “Jockusch's” denaturation temperature (determined as the temperature after which 50% of the protein is pelleted at pH 5.1 at 1500g) is 37-38°C.Fig. 2. Scanning rate and protein concentration dependences of TMV CP heat absorption. a) Scanning rate dependence of DeltaCp (scanning rates in °C/min are shown on the curves). Conditions: 30 mM PB, pH 8.0; protein concentration, 2 mg/ml. b) Concentration dependence of DeltaCp (protein concentrations in mg/ml are shown on the curves). Conditions: 30 mM PB, pH 8.0. Scanning rate, 1°C/min.
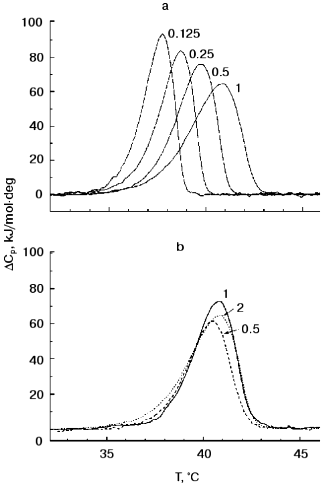
In contrast to Jockusch's test, in DSC experiments constant heating of samples is employed. The DSC heating regime most similar to that in the pH 5 test is 0.125°C/min. As seen in Fig. 2a, the TMV CP DSC melting temperature at this heating rate is 37.7°C, i.e., just the same as in the functional test. However, as will be shown below, a large part of the TMV CP secondary structure can be preserved even at much higher temperatures.
2. “Macroscopic” aggregation of tobacco mosaic virus coat protein. In the course of our studies of TMV CP thermal denaturation, we observed that on heating in a spectrophotometer cell of TMV CP solution in 50 mM PB, pH 7.0, a very large increase in turbidity (“absorption” at 313 nm) takes place [9, 10]. The spectral characteristics of this increase in absorption left no doubt that it is caused by the aggregation of the protein. For a 200-µg/ml TMV CP sample in a 1-cm cell, A313 reproducibly increased on heating from 0.02 to about 1.2 optical units and then dropped due to coagulation of the protein [9, 10]. We will call this process of formation of very large aggregates “macroscopic aggregation”.
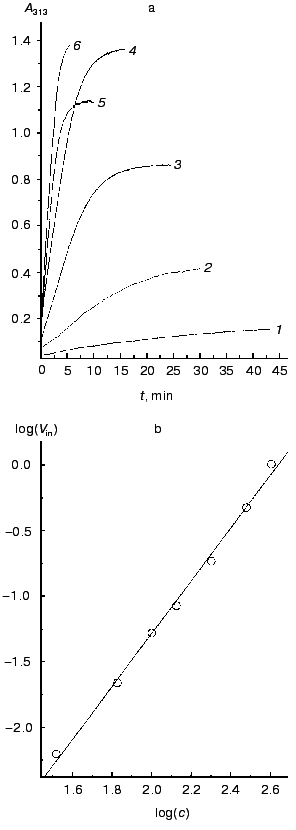
Figure 3a shows a time course of A313 increase on incubation at constant temperature of 52°C of different concentrations of TMV CP in 50 mM PB, pH 7.0. Aggregation can be observed at TMV CP concentrations as low as 33 µg/ml (2 µM). If other conditions (pH, ionic strength, temperature) remain constant, the final value of A313 attained is proportional to the CP concentration. This means that A313 values reflect the amount of aggregated protein. In all samples, after reaching the final A313 value, not less than 90% of the material could be pelleted by 1500g centrifugation; this shows that all (or almost all) TMV CP molecules in the samples are involved in the macroscopic aggregation. A similar picture was observed in 50 mM PB, pH 8.0.
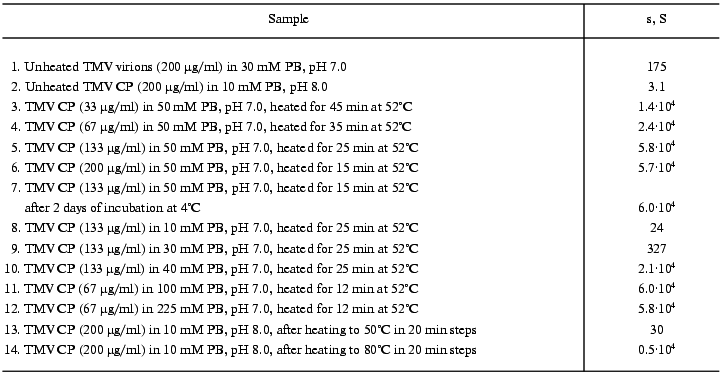
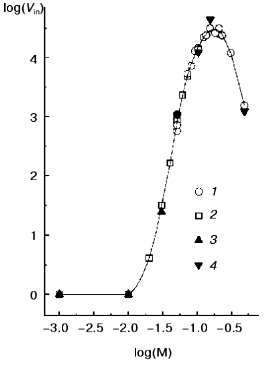
On reaching maximal A313 values, TMV CP samples were subjected to analytical centrifugation. As seen in the table (rows 3 to 6), 133 and 200 µg/ml samples of TMV CP in 50 mM PB, pH 7.0, have sedimentation coefficients (s) of (5-6)·104 S, while for 33 and 67 µg/ml those values were somewhat lower ((1.5-2.5)·104 S). Aggregates of (5-6)·104 S stay just on the verge of spontaneous precipitation, which occurs if samples are heated at 52°C for several more minutes. But at 4°C these aggregates remain stable in suspension for several days (rows 6 and 7 in the table). Aggregates of (1.5-2.5)·104 S are not yet ready for precipitation, and these samples may be further heated at 52°C for, say, 30 min, without any change in A313. Because the final A313 values (per concentration unit) were similar for all samples in Fig. 3a, in the 133 to 400 µg/ml range the final number of aggregates should be proportional to the initial CP concentration.Fig. 3. Concentration dependence of macroscopic aggregation of TMV CP. a) Turbidity (A313)-time profiles for different TMV CP concentrations. Protein concentrations (in µg/ml): 1) 33; 2) 67; 3) 133; 4) 200; 5) 300; 6) 400 (for 300 and 400 µg/ml, 0.5-cm cells were used). Conditions: 52°C, 50 mM PB, pH 7.0. b) The log-log dependence of the initial rate of aggregation (Vin, optical units/min) on TMV CP concentration (c, µg/ml).
Sedimentation coefficients of TMV CP aggregates
Note: Mean values from 3 to 10 experiments are presented. Heated samples
were centrifuged at 20°C after cooling in air to room
temperature.
The size of final (5-6)·10. S aggregates was estimated using the laser light-scattering method. The hydrodynamic radius (Rh) of the particles was found to be 117 nm as determined by cumulant analysis and 102 nm as determined by Tikhonov regularization [13] (mean value 110 nm). In the spherical approximation this corresponds to a volume of 5.6·106 nm3. In TMV virions, the CP subunit has a volume of 35 nm3 as calculated from the virion dimensions and the number of CP subunits per virion (2130). If we, in the absence of any other evidence, assume the same volume for the CP subunit in (5-6)·10. S aggregates, then on average one aggregate should contain about 1.6·105 subunits, i.e., 75 times more than the virion. At the same time, the sedimentation coefficient of the final aggregates is about 300 times higher than that of the TMV virion (180S). Even if we take into account the fact that TMV particles have very high axial ratio (17 : 1), the subunit packing density in macroscopic aggregates should be not lower than in TMV virions.
Figure 3b presents the log-log concentration dependence of initial rate (Vin) of A313 increase for heating TMV CP at 52°C in 50 mM PB, pH 7.0. In the range from 33 to 400 µg/ml, Vin was exactly proportional to the square of the TMV CP concentration (the inclination angle tangent is 2.02 and the correlation coefficient is 0.998). This may mean that TMV CP macroscopic aggregates grow by pair-wise collisions and fusion of preexisting smaller aggregates, and that the time of “double” aggregates formation is much shorter than the time between productive collisions.
We also found that TMV CP macroscopic aggregation displays very strong ionic strength dependence. The log-log dependence of the initial rate of aggregation at 52°C and pH 7.0 or 8.0 on phosphate buffer molarity is presented in Fig. 4 (for this buffer, ionic strength equals molarity multiplied by 2.4), and the final sedimentation coefficient values are given in rows 8 to 12 of the table. For 1 mM and 10 mM PB, pH 7.0 or 8.0, the initial value of A313 for 133 µg/ml TMV CP solution (0.02 optical unit) does not change at all within 22 min. In 20 mM PB, A313 increases in 22 min from 0.03 to 0.05, and in 30 mM PB from 0.04 to 0.10 optical unit. But, on further increase in buffer molarity, the initial rate of aggregation starts to rise very fast (Fig. 4). On going from 20 to 70 mM, the rate increases about 1300 times, and then the increase slows down. The maximal initial rate at both pH values is achieved at 150-200 mM, and then it starts to drop. Around 150 mM the process develops so fast that its rate cannot be measured at 133 µg/ml, and twice lower CP concentration should be used (in such cases the observed initial rates were multiplied by 4). Sedimentation coefficient values for samples with maximal A313 also rise very fast on increase in PB molarity from 30 to 40 mM and then level off at about 6·10. S (see table).
The observed character of aggregation rate dependence on ionic strength suggests that intermolecular hydrophobic interaction should be the main driving force of TMV CP macroscopic aggregation. As seen in Figs. 1 and 2, irreversible TMV CP denaturation occurs around 40°C and thus, like in many other cases [1, 2], TMV CP macroscopic aggregates should be formed by partially or fully denatured protein molecules. Because in DSC melting curves no additional endothermic or exothermic peaks were seen on further heating of TMV CP samples, this protein macroscopic aggregation is not accompanied by any cooperative heat-consuming or heat-producing processes.Fig. 4. Log-log ionic strength dependence of TMV CP macroscopic aggregation initial rate at pH 7.0 (1, 2) and 8.0 (3, 4). Protein concentration: 133 µg/ml CP (2, 3) and 67 µg/ml CP (1, 4); 67 µg/ml initial rate values were multiplied by 4. Measurements were done at 52°C.
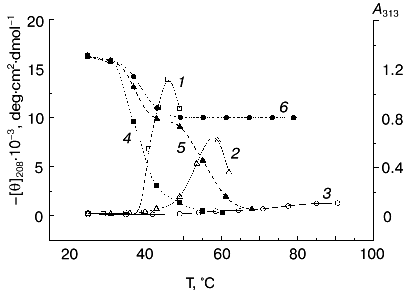
3. Structure of thermally denatured TMV CP. In further experiments, we studied the interrelation between the secondary structure of TMV CP and its macroscopic aggregation. Figure 5 (right ordinate) presents turbidity (A313) increase curves for heating of 200 µg/ml TMV CP in 10, 30, and 50 mM PB, pH 8.0, in the same regime (20 min heating in about 3°C steps) as in Jockusch's test [18, 19]. In accordance with the results of experiments at constant temperature of 52°C (Fig. 4), in this range an increase in the ionic strength results in a large decrease in aggregation temperature. In 50 mM PB, pH 8.0, aggregation temperature Tag (the temperature at which 50% of the total increase in A313 is observed) is 41°C, in 30 mM PB it rises to 52°C, and in 10 mM PB to ~73°C (Fig. 5). Decrease in ionic strength is also accompanied by decrease in maximal A313 value attained. In 30 and 50 mM PB, A313 quickly drops after reaching the maximal value due to the coagulation of the protein (Fig. 5).
Next, CD spectra in the 198 to 250 nm range were measured for TMV CP samples heated under identical conditions. Complete spectra are shown in Fig. 6, and the temperature dependence of [theta]208 is shown in Fig. 5 (left ordinate). At 25°C, TMV CP has a CD spectrum characteristic of alpha + beta proteins with high alpha-helix content, with a negative band at 208 nm and a shoulder at 222 nm ([theta]208 = -16,300 deg·cm2·dmol-1). Estimation of TMV CP alpha-helix content using the Greenfield-Fasman [20] equation (% alpha-helix = ([theta]208 - 4,000°)/29,000°) gives the value of 43%, in excellent agreement with the X-ray diffraction results [21, 22].Fig. 5. Temperature dependence of macroscopic aggregation and ellipticity at 208 nm for TMV CP at different PB (pH 8.0) molarities. Ordinate axes: 1-3) A313; 4-6) ellipticity. Conditions: 50 mM PB (1, 4); 30 mM PB (2, 5); 10 mM PB (3, 6). The protein samples (200 µg/ml) were heated in 20-min steps at the indicated temperatures.
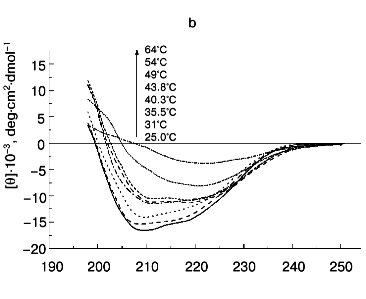
According to X-ray diffraction data [21, 22], the TMV CP subunit is built from two pairs of long, tightly packed, side-by-side alpha-helices that are connected by loops at both ends of the four-helix bundle. The protein contains high percent of alpha-helix (more than 45%) and only a small fraction of beta-structure (about 5%).Fig. 6. Temperature dependence of far-UV CD spectra of TMV CP at different PB (pH 8.0) molarities: a) 50 mM PB; b) 30 mM PB; c) 10 mM PB. The protein samples (200 µg/ml) were heated in 20-min steps at the indicated temperatures. The sequences of spectra at 208 nm and the indicated temperatures are shown on the figures.
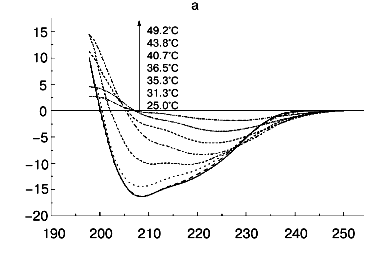
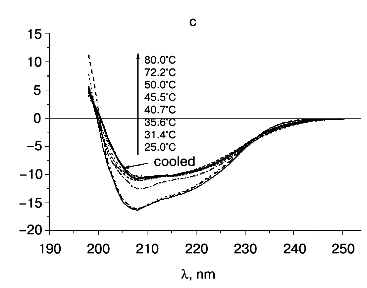
Step-wise heating of 200 µg/ml TMV CP in 50 mM PB, pH 8.0, to 34°C does not result in any changes in far-UV CD spectrum, but between 35 and 45°C a fast decrease in CD intensity, change in the spectrum shape, and a shift of the negative maximum from 208 to 222 nm are observed (Figs. 5 and 6a). Changes of this type are often seen during protein aggregation and are usually explained by an increase in the beta-structure content in aggregated proteins (see, for instance [1, 23]). At 55°C, the CD signal in the whole spectral region dropped almost to zero (Fig. 6a), presumably due to TMV CP precipitation (see Fig. 5).
Thus, in 50 mM PB, pH 8.0, TMV CP functional denaturation as revealed by the pH 5 test [10, 18, 19], heat transition (Figs. 1b), macroscopic aggregation (Fig. 5), and complete loss of the far UV CD signal (Figs. 5 and 6a) all occur in the same temperature interval (38-41°C). The coagulation of the protein under these conditions takes place at 49°C and thus cannot be the reason for most of the observed decrease in CD intensity.
A small decrease in PB molarity (from 50 to 30 mM) results in a drastic change in TMV CP CD melting (Figs. 5 and 6b). The initial decrease in [theta]208 occurs at a temperature close to that in 50 mM buffer, but after reaching a value of about -10,000 deg·cm2·dmol-1 (at ~42°C), the CD intensity stops to decrease and remains constant up to about 50°C. This means that in the temperature interval of about 40 to 50°C, TMV CP exists in a partially unfolded form (PUF). Then the ellipticity begins to drop again at temperatures coinciding with the start of macroscopic aggregation (Fig. 5). Ellipticity decreases almost to zero in parallel with turbidity increase, suggesting that the drop is driven by macroscopic aggregation.
This suggestion is confirmed by the results of TMV CP CD melting in 10 mM PB, pH 8.0 (Figs 5 and 6c). As seen in Fig. 5, in 10 mM buffer the turbidity increase is rather small, occurs only near 70°C, and is not followed by coagulation. Under these conditions, TMV CP is converted into PUF at nearly the same temperatures as in 30 mM PB, but it remains in this state upon heating up to at least 80°C (Figs. 5 and 6c).
The transition into PUF is not reversed by cooling of a TMV CP sample from 50°C to room temperature (Fig. 6c). The sedimentation coefficient of a 200 µg/ml sample cooled from 50 to 25°C was found to be about 30S, and that of a sample cooled from 80°C, about 5000S (table, lines 13 and 14), this suggesting a highly aggregated state of TMV CP subunits after heating to 50°C and especially to 80°C even in 10 mM PB.
To determine the state of aromatic amino acid residues in denatured TMV CP, we measured CD melting curves in the “aromatic” (250-300 nm) spectral region for 200 µg/ml TMV CP in 10 mM PB, pH 8.0. Between 37 and 40°C, i.e., at the same temperatures as in other denaturation tests (see above), TMV CP was found to completely and irreversibly lose optical activity in that spectral region (data not shown). This means that, in contrast to a muscle actin aggregating intermediate [23], in TMV CP a partially disordered form of aromatic amino acid residues lose the specific asymmetrical structure characteristic for the native protein.
DISCUSSION
As said above, according to X-ray diffraction data [21, 22] the main element of the TMV CP subunit (158 amino acid residues) structure is a four-helix bundle built from two pairs of alpha-helices (residues 19-33 and 37-52, and residues 73-86 and 111-135). Calculations based on the Greenfield-Fasman equation [20] show that, after transition from native ([theta]208 = -16,300 deg·cm2·dmol-1) form to PUF, TMV CP subunits retain about half (22%) of the initial alpha-helix content. It can be suggested that it is the longer, second pair of alpha-helices (73-86, 111-135) that is not broken in the PUF. The PUF is highly stable--in 10 mM PB it retains its secondary structure up to at least 80°C. Analytical centrifugation has shown that even after heating in 10 mM PB and to only 50°C, TMV CP subunits are highly aggregated (table, row 13).
Formation of a different type of ordered aggregates is one of the main functions of TMV CP [7, 8]. To fulfill it, the protein subunit bears large hydrophobic patches and charge clusters on its external surfaces that are responsible for axial and lateral inter-subunit interactions during polymerization [21, 22]. Possibly, the inter-helix interaction potential of TMV CP alpha-helices (or their pairs) is so high that, at neutral pH and moderate ionic strength, the protein cannot exist in the monomeric state and immediately forms different polymers (including virions) or produces smaller or larger “nonspecific” aggregates. And it is this potential that stabilizes the PUF and makes TMV CP thermal denaturation irreversible.
The results of melting experiments show that small (<=4S) aggregate forms are the most unstable state of TMV CP. In virions and in helical RP on one hand [14, 15] and in the partially structured PUF on the other (Fig. 6c), TMV CP is characterized by rather high thermal stability.
Fink suggested [1] that macroscopic protein aggregates are formed due to specific intermolecular interactions of hydrophobic surfaces of protein molecule subdomains, which are normally involved in tertiary intramolecular interactions between the same surfaces within one molecule. In the case of TMV CP, inter-subunit interactions responsible for normal ordered aggregation (polymerization) should possibly be also included. These interactions between individual alpha-helices or between their pairs stabilize the partially structured form of TMV CP in 10 or 30 mM PB, pH 8.0, against thermal unfolding. In 10 mM PB, macroscopic aggregation of the protein is restricted to about 30S aggregates at 50°C and to about 5000S aggregates at 80°C by electrostatic repulsion between subunits. Increase in PB molarity abolishes this repulsion and results in very large ((5-6)·104 S) aggregate formation followed by precipitation. During the formation of these very large aggregates (at about 40°C in 50 mM PB and at about 55°C in 30 mM PB), a change in the shape of the far-UV CD spectrum is observed, including loss of total intensity and shift of the negative maximum from 208 to about 220 nm (Fig. 6, a and b). This type of change in the CD spectra of aggregating protein is usually interpreted as a transition to beta-structure (see, for instance [1, 23]).
Changes in the protein and buffer concentrations exert almost no effect on TMV CP 4S aggregate thermal denaturation (Figs. 1, 2, 6), but strongly accelerate the macroscopic aggregation of the protein (Figs. 3-5). In 50 mM PB, pH 8.0, aggregation temperature exceeds denaturation temperature by only 2 to 4°C, and in 30 mM PB this difference increases to about 15°C. At first glance, this seems to contradict Fink's idea that some specific partly folded form of a protein is responsible for macroscopic aggregation [1]. But if we assume that for TMV CP it is the PUF that forms macroscopic aggregates, this contradiction can be avoided. The PUF is highly stable (Fig. 6c), and it can give a start to macroscopic aggregate formation in rather diverse conditions as soon as other factors (temperature, ionic strength, protein concentration) allow.
As seen in Figs. 1 and 2 (see also [15]), the TMV CP heat transition traces show practically no denaturational increment of heat capacity. This means that the extent of solvent-exposed protein subunit total hydrophobicity does not change significantly on thermal denaturation. Thus, the TMV CP transition to macroscopic aggregation should be determined not by an increase in the subunit total hydrophobicity but by surface exposition of some specific elements of the protein secondary structure during denaturation.
In some respects, our results resemble those reported by Uversky and coworkers [23] for the aggregation of an unfolding intermediate of another polymer-forming protein, muscle actin. The difference between the two proteins is, first, the absence of fixed asymmetrical structure of aromatic residues in the case of the TMV CP aggregating intermediate. It should also be said that in the case of actin the aggregation is restricted to relatively small size aggregates (6-15 monomers), while in the case of TMV CP very large aggregates (reaching many thousands of subunits) can be produced. The TMV CP aggregating intermediate can be considered as a border case between a highly ordered aggregate-producing form of actin [23] and the more or less fully disordered aggregating intermediates of many other proteins.
In conclusion, it should be said that the possibility to regulate temperature and extent of TMV CP “nonspecific” aggregation by small variations in buffer molarity or protein concentration may make this protein a rather useful model in studies of different aspects of macroscopic aggregation of proteins. Recently, on the basis of results of their structural study of an aberrant TMV CP ordered aggregate (so-called “stacked disk”), Caspar and Diaz-Avalos suggested the existence of an “amyloid-forming potential” in this protein [24].
This work was supported by the Russian Foundation for Basic Research (grants 99-04-48999 and 99-04-49133).
REFERENCES
1.Fink, A. L. (1998) Folding and Design,
3, R9-R23.
2.Kurganov, B. I. (1998) Biochemistry
(Moscow), 63, 430-432.
3.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci.
USA, 95, 13363-13383.
4.Horiuchi, M., and Caughey, B. (1999)
Structure, 7, R231-R240.
5.Georgalis, Y., Starikov, E. B., Hollenbach, B.,
Lurz, R., Schresinger, E., Saenger, W., Lehrach, H., and Wanker, E. E.
(1998) Proc. Natl. Acad. Sci. USA, 95, 6118-6121.
6.Wetzel, R. (1992) in Stability of Protein
Pharmaceuticals. Pt. B. In vivo Pathways of Degradation and
Strategies for Protein Stabilization (Ahern, T. J., and Manning, M.
C., eds.) Vol. 3, Plenum, N. Y., pp. 43-88.
7.Butler, P. J. G. (1984) J. Gen. Virol.,
65, 253-279.
8.Bloomer, A. C., and Butler, P. J. G. (1986) in
The Plant Viruses (van Regenmortel, M. H. V., and
Fraenkel-Conrat, H., eds.) Vol. 2, Plenum, N. Y., pp. 19-57.
9.Abu-Eid, M., Kust, S. V., Makeeva, I. V., Novikov,
V. K., and Dobrov, E. N. (1994) Molek. Genet. Mikrobiol.
Virusol., 3, 28-32.
10.Dobrov, E. N., Abu-Eid, M. M., Solovyev, A. G.,
Kust, S. V., and Novikov, V. K. (1997) J. Prot. Chem.,
16, 27-36.
11.Fraenkel-Conrat, H. (1957) Virology,
1, 1-4.
12.Dobrov, E. N., Kust, S. V., Yakovleva, O. A., and
Tikhonenko, T. I. (1977) Biochim. Biophys. Acta, 475,
623-637.
13.Koppel, D. E. (1972) J. Chem. Phys.,
57, 4814-4820.
14.Mutombo, K., Michels, B., Ott, H., Cerf, R., and
Witz, J. (1992) Eur. J. Biophys., 21, 77-83.
15.Orlov, V. N., Kust, S. V., Kalmykov, P. V.,
Krivosheev, V. P., Dobrov, E. N., and Drachev, V. A. (1998) FEBS
Lett., 433, 307-311.
16.Sanchez-Ruiz, J. M. (1992) Biophys. J.,
61, 921-935.
17.Kurganov, B. I., Lyubarev, A. E., Sanchez-Ruiz,
J. M., and Shnyrov, V. L. (1997) Biophys. Chem., 69,
125-135.
18.Jockusch, H. (1966) Z. Vererbungsl.,
98, 344-362.
19.Jockusch, H., Koberstein, R., and Jaenicke, R.
(1969) Z. Naturforsch., 24b, 613-617.
20.Greenfield, N., and Fasman, G. D. (1969)
Biochemistry, 12, 1290-1299.
21.Namba, K., Pattanayek, R., and Subbs, G. (1989)
J. Mol. Biol., 208, 307-325.
22.Bhyravbhatla, B., Watowich, S. J., and Caspar, D.
L. D. (1998) Biophys. J., 74, 604-615.
23.Kuznetsova, I. M., Biktashev, A. G., Khaitlina,
S. Yu., Vassilenko, K. S., Turoverov, K. K., and Uversky, V. N. (1999)
Biophys. J., 77, 2788-2800.
24.Diaz-Avalos, R., and Caspar, D. L. D. (2000)
J. Mol. Biol., 297, 67-72.