Transformation of a Fragment of beta-Structural Bacteriophage T4 Adhesin to Stable alpha-Helical Trimer
K. A. Miroshnikov1,2, N. V. Sernova2, M. M. Shneider2, and V. V. Mesyanzhinov2*
1Bach Institute of Biochemistry, Russian Academy of Sciences, Leninsky pr. 33, Moscow, 117071 Russia2Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 336-6022; E-mail: vvm@mail.ibch.ru
* To whom correspondence should be addressed.
Received April 18, 2000; Revision received August 21, 2000
Gene product 12 of bacteriophage T4, adhesin, serves to adhere the virus to host cells. Adhesin is a fibrous homotrimer, and a novel tertiary structure element, a beta-helix, is supposed to be a major structural feature of this protein. We have constructed two truncated gp12 mutants, 12N1 and 12N2, containing 221 and 135 N-terminal residues, respectively. When expressed in E. coli cells, these gp12 fragments formed labile beta-structural trimers. Another hybrid protein, 12FN, containing 179 N-terminal amino acid residues of gp12 fused to the C-terminal domain (31 amino acids) of T4 fibritin, was shown to have a trimeric proteolytically resistant alpha-helical structure. This structure is probably similar to that of fibritin, which has a triple alpha-helical coiled-coil structure. Hence, we have demonstrated the possibility of global transformation of fibrous protein structure using fusion with a C-terminal domain that initiates trimerization.
KEY WORDS: bacteriophage T4, protein folding, adhesin, fibritin, protein engineering, fibrous proteins
Abbreviations: PMSF) o-phenylmethylsulfonyl fluoride; SDS) sodium dodecyl sulfate; IPTG) isopropyl beta-D-thiogalactopyranoside; PCR) polymerase chain reaction; gp) gene product.
The process of polypeptide chain folding into a biologically active
spatial protein structure is often referred as “the second part of
the genetic code” [1]. It has been shown that
certain regions of amino acid sequence, and even individual residues,
can be crucial for initiation of protein tertiary structure formation
[2]. Folding of many proteins involves chaperones
[3], and this is the reason why the expression of
recombinant proteins in a foreign organism lacking specific chaperones
often results in misfolding and formation of inclusion bodies.
Misfolding caused by mutations is the main reason for heritable human
diseases, such as neurodegenerative disorders like Alzheimer's disease
or infectious prion formation [4, 5].
Fibrous proteins serve as a convenient model for studies of folding mechanisms because of their repetitive structure, which makes it possible to work with their fragments. Many fibrous proteins possess a C-terminal domain that initiates post-translational oligomerization. Such domains have been found in bacteriophage T4 fibritin [6], myosin, paramyosin [7], and reoviral adhesin [8]. We have solved the structure of a fibritin C-terminal domain, named foldon [9], by X-ray analysis [10]. Short tail fiber, gp12, another fibrous protein of bacteriophage T4, contains 526 amino acid residues per polypeptide chain, and it forms a homotrimer [11, 12]. Short tail fibers provide irreversible viral adsorption onto host cells [11-14]. Subsequent reorganization of a baseplate essential for tail sheath contraction and DNA injection into the cell also involves gp12 [15]. Our interest in this adhesin is based on the suggestion that its structure contains a novel beta-helical motif [16], and that folding of gp12 is assisted by the T4 phage chaperone gp57A [17, 18]. A sheer part of the gp12 polypeptide chain (residues 48-320) consists of six tandem repeats (40 residues in each) and additionally two shorter single repeats [16]. The gp12 repeats show a noticeable homology with phage T4 long tail fiber proteins, gp34 and gp37 [16, 19]. The chaperone gp57A also participates in the folding of gp34 and gp37 [20].
The length of full-size adhesin is about 32 nm [16], and the mature molecule probably has high asymmetry, so our attempts to produce crystals of whole adhesin have failed so far. Hence, we proposed construction and subsequent structural investigation of deletion fragments of gp12, as has been done previously for phage T4 fibritin. In our work, we constructed two fragments of gp12, shortened from the C-terminus. These fragments, denoted gp12N1 and gp12N2, contain 221 and 135 N-terminal amino acid residues of the protein monomer, respectively. Being expressed in E. coli, gp12 fragments formed labile beta-structural trimers. Co-expression of gp12 fragments with the chaperone gp57A did not change their properties (Miroshnikov and Shneider, unpublished results). However, chimeric protein 12FN, consisting of an adhesin fragment (179 N-terminal amino acid residues) and a phage T4 fibritin fragment (31 amino acid residues from foldon), showed unusual properties. Fusing a foldon to the C-teminus of the gp12 fragment resulted in the conversion of the beta-structural protein into a stable alpha-helical trimer.
MATERIALS AND METHODS
Engineering of gene constructions. DNA fragments were amplified by PCR according to a standard procedure [21]. T4 phage DNA was used as the PCR template. The sequences of PCR primers used were:
NcoI-12-fwd
5´-AAC-ACC-ATG-GGT-AAT-AAT-ACA-TAT
BamHI-12N1-rev
´-GCC-TTT-GGA-TCC-TTA-AGT-AGA-AGA
BamHI-12N2-rev 5´-TGA-GGA-TCC-AGT-TTA-AAC-TCG
BamHI-12FN-rev 5´-GGC-AGC-TGT-GGA-TCC-AGA-AGG
SacI - 12-rev 5´-TTA-GAG-CTC-TCA-TTC-TTT-TAC
BamHI-foldon-fwd
5´-CAA-GCT-CTC-CAA-GGA-TCC-GGT
EcoRI-foldon-rev
5´-CCC-CTC-TCC-TGA-ATT-CCT-AAA-CGA-AC
Amplified DNA fragments were purified using a QiaEx kit (QiaGen, Germany). DNA fragments were cloned into pET23d(+) (Novagen, USA) vector under control of T7 promoter. Clone selection and DNA isolation were done according to standard methods [21]. Plasmid pS57 employed for expression of gp57A was constructed on the basis of pACYC184 vector (Novagen). This plasmid contains the CAT gene and provides chloramphenicol resistance to cells. DNA sequence was determined by the Sanger method.
Protein expression. E. coli BL21(DE3) cells infected by plasmid pET23/g12 were grown in 2×YT media with ampicillin (100 µg/ml) at 37°C to A600 = 0.6, then induced by addition of IPTG to final concentration 1 mM [22], and incubated for 4 h. To produce full-size soluble gp12, we co-transformed E. coli BL21(DE3) with plasmids pS57 and pET23/g12, and cell growth/induction was processed in 2×TY with 100 µg/ml ampicillin and 25 µg/ml chloramphenicol at 25°C. Cells were harvested by centrifugation at 3000g for 15 min at 4°C.
Protein purification. Pelleted E. coli cells were resuspended in 15 ml of 50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10% (w/v) sucrose, 10 mM EDTA, 5 mM EGTA, 1 mM beta-mercaptoethanol, and 1 mM PMSF. The cell suspension was treated with lysozyme and DNAse for 30 min at room temperature [9] and then sonicated (6 × 15 sec at 4°C, Techpan TD-20). The cell debris was pelleted at 30,000g for 15 min at 4°C (rotor Sorvall JA-20). The supernatant was applied to an anion-exchange column (Hi-Trap Q cartridge, 5 ml; Pharmacia Biotech, Sweden), and the protein was eluted with a linear gradient of 50-300 mM NaCl in 50 mM Tris-HCl, pH 7.5, and 10 mM EDTA. The protein of interest was eluted at 0.13-0.15 M NaCl. Fractions containing the protein were pooled, PMSF added to final concentration 1 mM, and the protein concentrated to ~0.5 mg/ml using an Ultra Free 10 (Millipore, USA) concentrator and stored at 4°C.
The purification procedure for protein 12FN was a modification of a procedure for phage T4 fibritin [9]. Streptomycin sulfate was added to cell extract to final concentration 3%, and the extract was kept for 20 min at 4°C, then centrifuged at 30,000g for 15 min at 4°C. After separation of nucleic acid, the protein was precipitated by addition of ammonium sulfate to 28% saturation and subsequent incubation for 8 h at 4°C; then the protein was pelleted by centrifugation at 30,000g for 15 min at 4°C. The precipitate was resuspended in 2-5 ml 10 mM Na-phosphate buffer (pH 7.2) and then applied to a hydroxyapatite column (CHT-II, 5 ml; BioRad, USA). Protein was eluted by a stepwise gradient of Na-phosphate concentration. The purified protein was precipitated by adding ammonium sulfate to 30% saturation, subsequent incubation for 8 h at 4°C, and then centrifugation at 30,000g for 15 min at 4°C. The protein precipitate was resuspended in 2 ml of 50 mM Tris-HCl, pH 7.5, and dialyzed against the same buffer and stored at 4°C. Protein concentration was determined by the Bradford method.
Determination of protein oligomerization. Protein oligomerizationwas estimated by SDS-PAGE but using sample buffer contained no SDS or other denaturing agents. The SDS-resistant protein migrated as a band with mobility closely corresponding to the calculated trimer mass. When electrophoresis results could not be evaluated (for instance, the trimer band was unclear), gel filtration on Toyopearl HW-55 (Toyo Soda, Japan) or Superose 12 (Pharmacia Biotech) was applied to determine protein mass and oligomerization ratio, respectively. A 1 ×_50-cm column was equilibrated with buffer containing 50 mM Tris-HCl, pH 7.5, and calibrated using gel-filtration standards (Pharmacia Biotech).
Proteolysis patch. Trypsin (bovine pancreas, TPCK treated, Serva, Germany) was added to the partially purified or pure protein in buffer containing 100 mM Tris-HCl, pH 6.0, 1 mM CaCl2 at ratios from 1 : 5 to 1 : 100 (w/w) and incubated at 37°C. The reaction was terminated by adding PMSF to final concentration 1 mM and SDS-containing electrophoresis buffer, then the mixture was boiled on a water bath. The resulting samples were analyzed by SDS-PAGE.
Circular dichroism spectroscopy. Circular dichroism (CD) spectra of proteins were recorded using a JASCO J-500 spectropolarimeter at room temperature using 0.05-cm cuvettes (Hellma). Protein samples with concentration 50-100 µg/ml were dialyzed against 50 mM Na-phosphate buffer, pH 7.2. The CONTIN program was used for evaluation of structural content [23].
RESULTS AND DISCUSSION
The routine PCR-amplification approach was used to construct gp12 N-terminal mutants. We obtained DNA fragments with sizes 677 and 403 base pairs encoding deletion mutants 12N1 and 12N2, respectively. The fragments were cloned into the pET-23d vector using NcoI/BamHI sites. After 4-h induction, production of these proteins in E. coli cells reached up to 30% of total cell protein. The proteins 12N1 and 12N2 containing 221 and 135 amino acids were soluble (Fig. 1b).
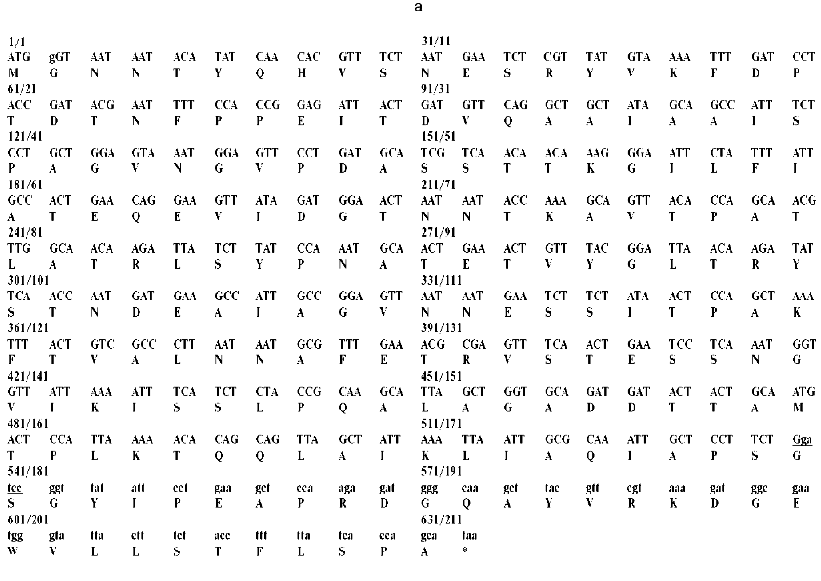
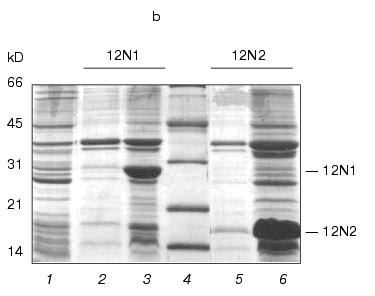
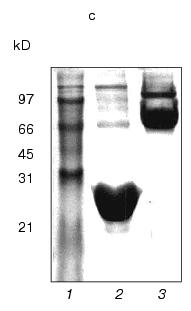
Fig. 1. a) Nucleotide and calculated amino acid sequence of the chimeric construction 12FN. The site of restriction endonuclease BamHI (GGATCC) is underlined. It was used to join the gene 12 fragment and the foldon. Small letters shows the nucleotide sequence of the fibritin foldon. The generation of the site NcoI (CCATGG) (not shown), containing initiation codon Met, resulted in a point mutation that changes Ser to Gly. The change of Glu to Gly is the result of the BamHI generation site. b) Expression of recombinant proteins 12N1 and 12N2 in E. coli Bl 21(De3) cells. The precipitates (lanes 2 and 5) and the supernatants (lanes 3 and 6) of lysed cells expressing 12N1 and 12N2, respectively, are presented. Lane 1, the lysate of E. coli cells before induction with IPTG; lane 4, BioRad protein marker (14, 21, 30, 45, and 66 kD). Cell extracts were analyzed in 15% polyacrylamide gel with 1% SDS. c) The stability of triple structure of the protein 12FN to SDS: 1) BioRad protein marker; 2) sample of denatured 12FN; 3) sample of native protein. Electrophoresis in 12% polyacrylamide gel with 2% SDS.Unlike many homotrimeric fibrous proteins, such as full-length gp12 [17], fibritin [24], phage P22 tailspike protein [25], and reovirus adhesin sigma1 [8], gp12 fragments revealed no stability to SDS. SDS-PAGE of cold-sampled proteins gp12N1 and gp12N2 showed no lower mobility band corresponding to the oligomer. However, the elution volumes on gel filtration of gp12N1 and 12N2 (Toyopearl HW-55 or Superose 12) suggested the molecular masses of the proteins to be about 75 and 50 kD. This serves as evidence of trimeric organization of both proteins. Gel filtration of the full-length gp12 showed a mass as 165 kD, which is very close to calculated value, 177 kD (data not shown).
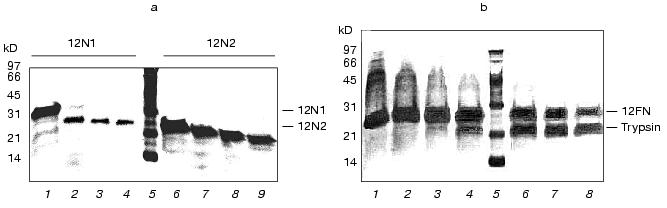
Trypsin treatment has that proteins 12N1 and especially 12N2 are stable to proteolysis (Fig. 2a). Gp12 protein was stable up to trypsin/protein ratio 1 : 20 (w/w), but at higher values it undergoes fast degradation into short fragments.
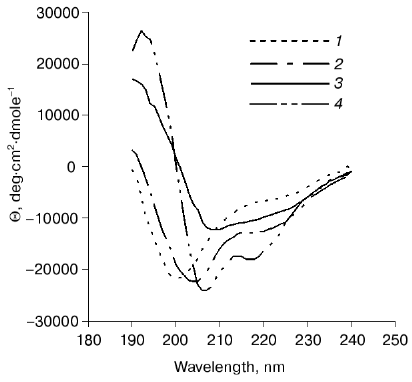
The circular dichroism spectra of proteins 12N1 and 12N2 are similar and have strong negative minimum of ellipticity at 205 nm (Fig. 3). Formal calculation according to CONTIN [23] shows that the secondary structures of the proteins are mixtures of beta-structure (32% for 12N2) and unordered structure. Such high content of structure which seems unordered is in contradiction with the fact of their trimer organization and their stability to proteolysis.Fig. 2. a) Proteolysis of proteins 12N1 and 12N2 with trypsin. E/S = 1 : 20 (w/w). Lanes 1-4 and 6-9 correspond to incubation with 1 mM CaCl2 at 37°C for 0, 5, 10, and 15 min for proteins 12N1 and 12N2, respectively; 5) BioRad protein marker. Electrophoresis in 15% polyacrylamide gel with 2% SDS. b) Proteolysis of protein 12FN with the trypsin. Incubation at 37°C for 15 min. E/S = 0 (1), 1 : 200 (2), 1 : 100 (3), 1 : 50 (4), 1 : 20 (6), 1 : 10 (7), 1 : 5 (8) (w/w); 5) BioRad protein marker. Electrophoresis in 15% polyacrylamide gel with 2% SDS.
Coexpression of gp12 and the chaperon gp57A gives us soluble full-sized gp12. Gp12 has a CD spectrum similar to that of proteins 12N1 and 12N2. It contains 28% alpha-, 31% beta-, and 41% unordered structure (Fig. 3). Interestingly, the only published beta-helical CD spectrum, that of Bordetella pertussis hemagglutinin [26], is specific for beta-sheet protein. But, it seems adhesin T4 trimer has a more complex structure, and we do not have enough data to interpret its CD spectrum. It is possible that the N-terminal deletion mutant structure is different from the native structure. It could be a “molten fiber”, which has some properties of a globular protein, but still retains trimeric organization.Fig. 3. CD spectra of proteins 12N1 (1), 12N2 (2), gp12 (3), and 12FN (4). The spectra were measured in 50 mM Na-phosphate buffer at room temperature.
We tried to stabilize gp12 fragments using the fibritin foldon. This C-terminal domain, containing 31 amino acids and having triple beta-propeller-like structure [9], initiates T4 phage fibritin trimerization, which forms an fibrous alpha-helical coiled-coil protein. We have demonstrated the possibility of construction of chimeric proteins in which a triple adhesin fragment is a C-terminal extension of shortened fibritin [27]. In this work, we constructed a chimeric protein joining a fibritin foldon to the adhesin C-terminus (Fig. 1a). Protein 12FN, containing 179 amino acids from gp12 and the fibritin foldon on the end, was totally soluble. It was purified like fibritin using fractionation with ammonium sulfate and chromatography on a hydroxyapatite column [22].
Protein 12FN is a stable trimer and, unlike 12N1 and 12N2, is SDS-tolerant, like fibritin (Fig. 1c). The mobility of unheated protein 12FN in SDS-PAGE is close to ~70 kD, this confirming its trimeric structure. Gel filtration of gp12FN on a calibrated Superose 12 column showed an apparent protein mass as 72 ± 2 kD, this being nearly equal to the calculated mass of a trimer of 69 kD (data not shown). Protein 12FN is more stable to proteolysis than 12N1 and 12N2. We did not observe protein fragmentation up to trypsin/protein ratio 1 : 5 (w/w) on incubation for 15 min at 37°C (Fig. 2b).
The circular dichroism spectrum of protein 12FN shows alpha-helical structure (Fig. 3) and has noticeable differences from the CD spectra of both deletion mutants (12N1 and 12N2) and gp12. Heptade repeats, which are characteristic for alpha-helical coiled-coil proteins, are absent from the gp12 sequence. Generally, super-helical formation of coiled-coil type is possible even without distinctive heptade periodicity [28]. So, we could not exclude that chimeric oligomer protein 12FN is like a super-helical fibritin.
In 1994, J. Rose and T. Creamer postulated the “Paracelsus challenge” principle [29], supposing that the type of protein structure totally changes when a 50% change of amino acid sequence takes place. Recently, experimental proof of this paradigm was demonstrated by conversion of beta-sheet structure protein to small alpha-helical RNA-binding protein [30]. In this case, a domain, which was intermediate between fragments, had tight homology with both proteins. This amino acid sequence of this domain was changed according to a calculated alpha-helix. From the other side, chimeric proteins, which contained domains from two different proteins and combined functional properties of both parental proteins, were described. A combinations of human lysozyme and alpha-lactalbumin for studying folding intermediates [31] and combination of three different Na+-nucleoside transport proteins in one [32] may be listed as an outstanding example of this approach. So, domain interaction was insignificant and did not change tertiary structure of each domain. All chimeric proteins mentioned above were constructed on the base of small monomer proteins that were able to refold spontaneously.
The possibility of global transformation of protein structure without significant changes of amino acid sequence was demonstrated by means of model peptides when amyloidogenesis and prions were studied [33]. Minimal mutagenesis, change of physical conditions [34], and the presence of organic solvents [35] result in irreversible transition of alpha-helix to beta-structure and vice versa. However, such dramatic changes in protein structure were never described for such comparatively large protein as 12FN. The fibritin foldon, being on the C-terminal of a chimeric protein, was able to direct folding de novo to alpha-helix. Proteins 12N1 and 12FN have 81% homology, 12N2 and 12FN have 62% homology, and, moreover, the polypeptide chain fragments from gp12 are completely identical. So, the example of the protein engineering that we described could be the common solution of the “Paracelsus challenge”. We are now crystallizing the chimeric protein 12FN to solve its X-ray structure.
This work was supported in part by grants of the Howard Hughes Medical Institute (75195-520803), the Russian Foundation for Basic Research (99-0448430), the program “Universities of Russia” (2608-415/098), and by a grant after Contest-Examination of Scientific Projects of Young Scientists of the Russian Academy of Sciences (resolution of the Presidium of the Russian Academy of Sciences No. 72 of July 13, 1998).
REFERENCES
1.Sobolev, B. N., Shneider, M. M., Marusich, E. I.,
and Mesyanzhinov, V. V. (1995) Evolutionary Biochemistry and Related
Areas of Physicochemical Biology (Poglazov et al., ed.), pp.
351-374.
2.King, J. (1989) Chemistry & Engineering
News. Special Report, 4, 32-54.
3.Georgeopulos, C. P., Hendrix, R. W., Casjens, S.
R., and Kaiser, A. D. (1973) J. Mol. Biol., 76,
45-60.
4.Cohen, F. E. (1999) J. Mol. Biol.,
293, 313-320.
5.Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M.,
Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R. J.,
Cohen, F. E., and Pruisner, S. B. (1993) Proc. Natl. Acad. Sci.
USA, 90, 10962-10966.
6.Strelkov, S. V., Tao, Y., Shneider, M. M.,
Mesyanzhinov, V. V., and Rossmann, M. G. (1998) Acta Crystallogr. D.
Biol. Crystallogr., 54, 805-816.
7.Cohen, C., and Parry, D. A. D. (1998) J. Struct.
Biol., 122, 180-187.
8.Gilmore, R., Coffey, M. C., Leone, G., McLure, K.,
and Lee, P. W. (1996) EMBO J., 15, 2651-2658.
9.Efimov, V. P., Nepluev, I. V., Sobolev, B. N.,
Zurabishvili, T. G., Schulthess, T., Lustig, A., Engel, J., Haener, M.,
Aebi, U., Ven'yaminov, S. Yu., Potekhin, S. A., and Mesyanzhinov, V. V.
(1994) J. Mol. Biol., 242, 470-486.
10.Tao, Y., Strelkov, S. V., Mesyanzhinov, V. V.,
and Rossmann, M. G. (1997) Structure, 5, 789-798.
11.Mason, V. S., and Haselkorn, R. (1972) J. Mol.
Biol., 66, 445-469.
12.Dickson, R. C. (1970) J. Mol. Biol.,
53, 633-647.
13.Zorzopulos, J., Kozloff, L. M., Chapman, V., and
DeLong, X. (1979) J. Bacteriol., 137, 545-555.
14.Crowther, R. A., Zenk, E. V., Kikushi, Y., and
King, J. (1977) J. Mol. Biol., 116, 489-523.
15.Selivanov, N. A., Prilipov, A. G., Efimov, V. P.,
Marusich, E. I., and Mesyanzhinov, V. V. (1990) Biomed.
Sci.,1, 55-62.
16.Makhov, A. M., Trus, B. L., Conway, J. F., Simon,
M. N., Zurabishvili, T. G., Mesyanzhinov, V. V., and Steven, A. C.
(1993) Virology, 194, 117-127.
17.Curits, T. S., Selivanov, N. A., and
Mesyanzhinov, V. V. (1984) Dokl. AN SSSR, 274,
448-452.
18.Burda, M. R., and Miller, S. (1999) Eur. J.
Biochem., 265, 771-778.
19.Selivanov, N. A., Prilipov, A. G., Efimov, V. P.,
Marusich, E. I., and Mesyanzhinov, V. V. (1990) Biomed. Sci.,
1, 55-62.
20.Cerritelli, M. E., Wall, J. S., Simon, M. N.,
Conway, J. F., and Steven, A. C. (1996) J. Mol. Biol.,
260, 767-780.
21.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold
Spring Harbor Press.
22.Studier, F. W., Rosenberg, A. N., Dunn, A. H.,
and Dubendorff, J. W. (1990) Meth. Enzymol., 185,
60-69.
23.Provencher, S. W., and Glockner, J. (1981)
Biochemistry, 20, 33-37.
24.Sobolev, B. N., and Mesyanzhinov, V. V (1991)
J. Biom. Str. Dynamics, 8, 953-965.
25.Steinbacher, S., Seckler, R., Miller, S., Steipe,
B., Huber, R., and Reinemer, P. (1994) Science, 265,
383-386.
26.Makhov, A. M., Hannah, J. H., Brennan, M. J.,
Trus, B. L., Kocsis, E., Conway, J. F., Wingfield, P. T., Simon, M. N.,
and Steven, A. C. (1994) J. Mol. Biol., 241, 110-124.
27.Miroshnikov, K. A., Marusich, E. I., Cerritelli,
M. E., Cheng, N., Hyde, C. C., Steven, A. C., and Mesyanzhinov, V. V.
(1998) Protein Eng., 11, 97-104.
28.Hicks, M. R., Holberton, D. V., Kowalczyk, C.,
and Woolfson, D. N. (1997) Folding & Design, 2,
149-158.
29.Rose, J. D., and Creamer, T. P. (1994)
Proteins: Struct. Funct. Genet., 19, 1-3.
30.Dalal, S., Balasurbramanian, S., and Regan, L.
(1997) Nature Struct. Biol., 4, 548-552.
31.Van Dael, H. (1998) Cell. Mol. Life Sci.,
54, 1217-1230.
32.Wang, J., and Giacomini, K. M. (1999) Mol.
Pharmacol., 55, 234-240.
33.Mihara, H., Takahashi, Y., and Ueno, A. (1998)
Biopolymers, 47, 83-92.
34.Zhang, S., and Rich, A. (1997) Proc. Natl.
Acad. Sci. USA, 94, 23-28.
35.Ragona, L., Confalonieri, L., Zetta, L., DeKruif,
K. G., Mammi, S., Peggion, E., Longhi, R., and Molinari, H. (1999)
Biopolymers, 49, 441-450.