Expression, Refolding, and Ferritin-Binding Activity of the Isolated VL-Domain of Monoclonal Antibody F11
A. P. Dubnovitsky1, Z. I. Kravchuk1, A. A. Chumanevich1, A. Cozzi2, P. Arosio2, and S. P. Martsev1*
1Institute of Bioorganic Chemistry, National Academy of Sciences of Belarus, ul. Akademika Kuprevicha 5/2, Minsk, 220141 Belarus; fax: +375-17-2-63-7274; E-mail: martsev@ns.iboch.ac.by2DIBIT, Scientific Institute H. San Raffaele, Milano, Italy
* To whom correspondence should be addressed.
Received April 14, 2000
Expression of the VL-domain of mouse monoclonal antibody F11 to human spleen ferritin in Escherichia coli cells is associated with the formation of insoluble protein aggregates (inclusion bodies). The aggregates were solubilized in the presence of guanidine hydrochloride and the recombinant VL-domain was purified by immobilized metal affinity chromatography (IMAC). Subsequent renaturation results in ~99% pure preparation with high yield. The VL-domain forms dimers at concentrations from 1 to 10 mg/ml. Monomeric form is detected only at protein concentrations below 0.5 mg/ml. Functional activity of the VL-domain was verified by two variants of ELISA. The affinity of the VL-domain ((0.2-1.2)·108 M-1) is similar to the affinity of the full-length parental antibody F11 because when the immobilized VL-domain was used, the binding constant of ferritin to the VL-domain was only 4-6-fold lower than that in the case of F11 antibody. In another ELISA system with immobilized ferritin, affinity was decreased 30-fold. The VL-domain of antibody F11 is the first example of the recombinant variable domain of the immunoglobulin light chain that preserves the antigen-binding activity in the absence of the partner VH-domain. The data indicate that the recombinant VL-domain can be used in construction of chimeric immunotoxins and other antigen-binding proteins in immunotherapy and in studies of correlations between folding, stability, and activity of immunoglobulins.
KEY WORDS: VL-domain, recombinant antibody, immunoglobulin domain, folding, protein expression, antigen binding
Abbreviations: IMAC) immobilized metal affinity chromatography; ELISA) enzyme-linked immunosorbent assay; IPTG) isopropyl beta-D-thiogalactopyranoside; scFv) single-chain Fv-fragment; FPLC) fast protein liquid chromatography; PBS-BSA) sodium phosphate buffer containing 1% bovine serum albumin.
Recombinant antigen-binding fragments of immunoglobulins are now
considered as potential transport modules for targeted immunotherapy of
cancer and related diseases. Variable domains VL and VH of the light
and heavy chains combine in the Fv-fragment of the antibody molecule
and participate in the formation of the antigen-binding site of the
antibody. The two-domain single-chain antibodies, also designated
scFv-fragments, are the minimal structure preserving the complete
antigen-binding site [1, 2].
Unlike the Fv-fragments of the full-length antibodies, in the
scFv-fragments the VH- and VL-domains are covalently bound by a linker
polypeptide of variable length. On the other hand, single domain
constructs were recently used as functional immunoglobulin analogs [3-6]. Single domain fragments have
low molecular weight (~14 kD) and are thus better expressed in
bacteria; they have lower potential immunogenicity in vivo and
better penetrate into the target cells. Decreased or even lost
antigen-binding activity (due to the absence of the partner domain in
the incomplete construct of the active site) can be restored by genetic
manipulations [3, 4].
The VH-domain significantly contributes to the formation of the antigen-binding site [7] and is considered as a more promising (versus the VL-domain) structure for construction of immunotoxins. On the other hand, the disadvantages of the VH-domain include its poor solubility and its aggregation. This requires increasing solubility of the VH-domain by methods of genetic engineering, for example, by camelization, i.e., site-directed mutagenesis to confer the structural properties of the VH-domains of camelides, whose VH-domains have unusually high solubility [4, 5].
The VL-domain has not been extensively studied as an independent basis for immunotoxin construction. Advantages of the VL-domain include its high solubility in all organisms and its resistance to aggregation, which may be associated with the formation of the VL-VL homodimer. A VL-domain-based immunotoxin has higher antigen-binding activity than that of cytotoxic VH-domain derivatives [8]. The study of Brinkmann et al. [8] is, to our knowledge, the only experimental evidence of the advantages of the VL-domain versus the VH-domain in antigen binding and construction of immunotoxins.
Isolated immunoglobulin domains and their two-domain variants are of interest also because these structures are the simplified models of immunoglobulin folding analysis; construction analysis of full-length multi-domain antibodies is not yet feasible. Additional interest in the VL-domains is due to their direct involvement in pathological amyloidogenesis. This domain is critical for the formation of amyloid fibrils from the light immunoglobulin chains and the Bence-Jones proteins [9-11].
We recently described the preparation, folding, and stability of the functionally active recombinant scFv-fragment [12] derived from the mouse monoclonal antibody F11 to human spleen ferritin [13-15]. In the present work, preparation and properties of a derivative of this construct, the VL-domain, is described as this is the first example of a recombinant VL-domain which preserves its antigen-binding function. We also show that the functional VL-domain forms a more open binding site with less steric hindrances than the parental full-length antibody F11; unlike F11, it can bind conformational variants of the antigen with the same affinity. The data indicate that the VL-domain can be used to construct chimeric immunotoxins and other antigen-binding proteins for clinical treatment, and it is an interesting model for studies of the relationship mechanisms between folding, stability, and activity in antibodies.
MATERIALS AND METHODS
Construction of the VL-expressing plasmid pETVL. The pETVL plasmid encodes for the isolated VL-domain of the mouse monoclonal antibody F11 to human spleen ferritin [13]; the plasmid was generated by the standard methods from the pETscF11 plasmid encoding for the single-chain scF11 antibody composed of the VH-domain bound via a linker peptide with the VL-domain [16]. The VL-domain-encoding fragment was amplified by the polymerase chain reaction from the pETscF11 plasmid DNA using direct primer (5´-GCCGCCCATATGGACATCCAGATGACTCAGTCT-3´) including the NdeI restriction site and reverse primer (5´-GCCGCCGGATCCCTAGTGGTGGTGGTGGTGGTGCCGTTTTATTTCCAACTTTGT-3´) to add the BamHI restriction site and sequence of six histidine residues to the C-terminus of the VL-domain. The thus synthesized gene of the VL-domain was cleaved by NdeI and BamHI and inserted in a NdeI/BamHI-cleaved pET12b plasmid under the control of the T7 RNA-polymerase promotor. Then the reaction mixture was used to transform Escherichia coli cells (strain BL21(DE3)pLysS). Thus, the pETVL plasmid included the VL-domain-encoding sequence containing an additional Met residue at the N-terminus. Moreover, the C-terminal (His)6-tag was substituted for the Arg residue present at the C-terminus of the VL-domain of the parental full-length antibody F11.
Expression and purification of the recombinant VL-domain. The Escherichia coli cells (strain BL21(DE3)pLysS) were transformed with the pETVL plasmid under standard conditions [17]. Expression was performed in conical flasks with mixing in 1 liter of LB-medium [17] containing ampicillin (0.1 g/liter) and chloramphenicol (0.17 g/liter). The cells were grown at 37°C. Overnight culture was added at 1 : 100 dilution. Expression of the recombinant VL-domain gene was induced by addition of 10 µM isopropyl beta-D-thiogalactopyranoside (IPTG) when the optical density of the culture at 550 nm reached 0.5. After 3.5-4.0 h of incubation of the culture at 30°C, the cells were pelleted by centrifugation at 3000g for 15 min at 4°C. The pellet of the cells was frozen and stored at -10°C.
Cells collected from 1 liter of culture were resuspended in 70 ml of 0.1 M Tris-HCl buffer (pH 8.1) and disintegrated by sonication. The suspension was centrifuged for 30 min at 40,000g in the cold to separate soluble cellular proteins, and the insoluble fraction (inclusion bodies) was washed three times with 0.1 M Tris-HCl buffer (pH 8.1); the pellet was solubilized in 30 ml of 6 M guanidine hydrochloride for 1 h with mixing. The mixture was centrifuged for 30 min at 40,000g, and the supernatant was dialyzed against 25 mM Tris-HCl buffer (pH 7.0) containing 6 M urea (buffer A). The mixture was centrifuged and the pellet was discarded; imidazole was added to the supernatant to 30 mM and the solution was loaded on a Ni-NTA-Sepharose column equilibrated with buffer A containing 50 mM imidazole. The column was washed with 5 volumes of the buffer, and the bound VL-domain was eluted with 1.5 volumes of buffer A containing 250 mM imidazole.
Refolding of the recombinant protein. The VL-domain-containing IMAC fractions were pooled and sequentially dialyzed against 50 mM Tris-HCl (pH 8.1) containing 3 M urea and 1 M urea. Subsequent protein refolding (protein concentration not exceeding 1 mg/ml) was performed by controlled dialysis. Sodium phosphate buffer (50 mM; pH 7.4) was substituted for the buffer containing 1 M urea (total volume 500 ml) at 50 ml/h flow rate. The refolding time was 48 h. Refolding yield at protein concentration 0.3-0.5 mg/ml was up to 100%.
Functional activity assay. The assay included two variants of competitive ELISA (two different forms of immobilized antigen) and noncompetitive ELISA using the immobilized VL-domain.
For competitive ELISA, 50 ng of monoclonal biotin-labeled F11 antibody and increasing amounts of the VL-domain or parental antibody (in 250 µl of 0.1 M sodium phosphate buffer, pH 7.4, containing 1% bovine serum albumin (PBS-BSA)) were added to human spleen ferritin immobilized directly on polystyrene or on rabbit polyclonal antibodies. After incubation for 2 h at 22°C with mixing and triple washing with distilled water, 250 ng of streptavidin-horseradish peroxidase conjugate (in PBS-BSA) were added to the samples. After 30 min, immobilized enzyme activity was determined by spectrophotometry [15] by registering the optical density at 492 nm after incubation at room temperature for 5 min. The substrate mixture contained 0.1% o-phenylenediamine and 0.05% H2O2 in 0.1% sodium citrate (pH 4.5). The reaction was terminated by addition of 10% sulfuric acid to the samples.
For noncompetitive ELISA, increasing amounts of ferritin in PBS-BSA were added to the tubes containing the immobilized recombinant VL-domain or parental monoclonal antibody; the samples were mixed at 22°C for 1.5 h. Then, immobilized immune complexes were stained with biotin-labeled rabbit polyclonal antibodies and streptavidin-peroxidase conjugate. Immobilized enzyme activity was determined as described above.
All ligand-binding assays were performed at room temperature in triplicate and the means were plotted. The association constants were determined using the saturation curves in double reciprocal plots. Variations in the association constant determination did not exceed 16%.
Gel-filtration. Gel-filtration was performed using a Pharmacia FPLC system (Pharmacia LKB Biotechnology, Sweden) on a Superose 12, HR 10/30 column (1.5 × 35 cm). Samples containing variable concentrations of the VL-domain were loaded in 100 µl of 50 mM sodium phosphate buffer (pH 7.4). The column was calibrated using molecular weight markers (Sigma, USA) including cytochrome c (12.3 kD), chymotrypsinogen (25 kD), ovalbumin (45 kD), bovine serum albumin (67 kD), immunoglobulin G (150 kD), and blue dextran (2000 kD).
Other methods. Biotin-labeled monoclonal and polyclonal antibodies were prepared as described [13].
The concentration of the VL-domain was determined by spectrophotometry (extinction coefficient A1cm,280 1% = 11.2 calculated as described [18]).
The recombinant protein was concentrated using a Millipore (USA) concentrator.
The composition of the cellular fractions and purity of the preparation were analyzed by SDS-polyacrylamide gel electrophoresis through 15% gel by the method of Laemmli [19]. The gels were stained with Coomassie brilliant blue R-250.
RESULTS
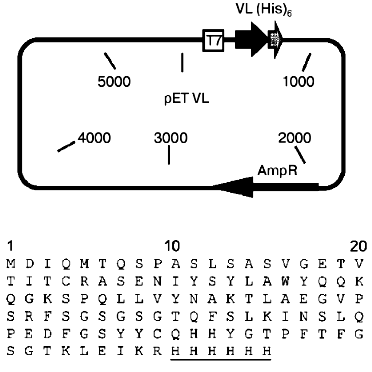
Plasmid construction. Based on the pET(scF11) plasmid encoding for the recombinant single-chain antibody to human spleen ferritin [16], the pETVL plasmid (Fig. 1) was constructed. The plasmid contains the gene of the isolated variable domain of the light chain of monoclonal antibody F11 [13] under the control of the IPTG-inducible promotor of T7 RNA-polymerase. A (His)6-tag was added to the C-terminus of the single-chain antibody because this is required for isolation of the recombinant protein by immobilized metal affinity chromatography [20, 21].
Expression and refolding of the recombinant VL-domain. The cells of E. coli (strain BL21(DE3)pLysS) transformed with pETVL plasmid are resistant to two antibiotics--ampicillin and chloramphenicol. The ampicillin-resistance gene is localized in the pETVL plasmid encoding for the recombinant VL-domain of monoclonal antibody F11. Apart from this plasmid, the cells of E. coli BL21(DE3)pLysS contain a pET-vector family-compatible plasmid pLysS which carries the chloramphenicol-resistance gene. This plasmid provides for the low level of expression of T7-lysozyme in the bacterial cells (natural inhibitor of T7 RNA-polymerase) thus suppressing the transcription of the T7-promotor in the absence of the inducer IPTG. When E. coli cells are induced with IPTG, the pLysS plasmid does not significantly influence the growth rate of the bacterial cells and expression of the target genes. Hence, the presence of the pLysS plasmid simplifies transformation of the E. coli BL21(DE3) cells with plasmids containing insertions toxic for the host (for example, the VL-domain gene) and increases the stability of these plasmids in the cell, providing for high expression level.Fig. 1. Construction of the plasmid pETVL carrying the recombinant VL-domain gene (upper panel) and its amino acid sequence (lower panel). AmpR, the ampicillin-resistance gene; T7, T7 RNA-polymerase promotor; VL, sequence encoding for the variable light chain domain of the parental monoclonal antibody F11; (His)6, C-terminal tag of six histidine residues (underlined).
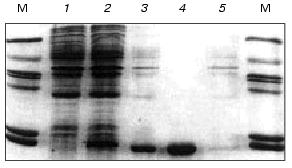
After induction with IPTG and expression of the VL-domain gene, the recombinant protein was accumulated in insoluble form as bacterial inclusion bodies. The level of expression was high, and the recombinant VL-domain comprised ~10% of the cellular protein (Fig. 2, lane 2). According to SDS-polyacrylamide gel electrophoresis of the ultrasound-disintegrated cell fraction, the inclusion bodies contained up to 70% of the target protein (Fig. 2, lane 3).
The data on the effect of cell culture growth temperature before and after induction indicate that after expression the amount of VL-domain was maximal at 28-32°C. Changes in the culture growth temperature before the induction influenced only the growth rate of the cells. Optimization of the temperature of the bacterial culture growth during the expression of the recombinant antibody fragment increased the yield of the VL-domain (40-60 mg per liter of bacterial culture) versus that described in the literature [22-24].Fig. 2. Expression and purification of the recombinant VL-domain: 1, 2) E. coli cells before and after induction, respectively; 3) soluble fraction of inclusion bodies in 6 M urea; 4) fraction containing VL-domain after IMAC; 5) contaminating protein not bound to the sorbent; M, molecular weight markers including bovine serum albumin (66 kD), ovalbumin (45 kD), histone H1 subfraction (36 and 34 kD), carbonic anhydrase (29 kD), alpha-lactalbumin (14 kD), and cytochrome c (13 kD).
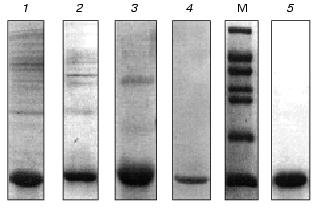
After solubilization of the inclusion bodies with guanidine hydrochloride, the recombinant VL-domain was purified by immobilized metal affinity chromatography under denaturing conditions (in the presence of 6 M urea) to prevent the formation of insoluble protein aggregates. The soluble fraction of the inclusion bodies was transferred from guanidine hydrochloride into 25 mM Tris-HCl buffer (pH 7.0) containing 6 M urea. Then the solution was loaded on a Ni-NTA-Sepharose column. The VL-domain was bound to the beads due to the (His)6-tag on the C-terminus of the molecule, and contaminating proteins did not bind to the sorbent (Fig. 2, lane 5). After washing and exchanging the buffer, subsequent elution yielded the VL-domain preparation with purity over 90% (Fig. 2, lane 4). Subsequent dialysis against buffers containing 3 M and 1 M urea was associated with significant precipitation of residual contaminating proteins with lower solubility than that of the VL-domain. The recombinant protein remained in solution. During the last refolding stage, residual contaminating proteins and nucleic acids were removed, and according to SDS-polyacrylamide gel electrophoresis, the purity of the VL-domain exceeded 95% (Fig. 3, lane 5).
In addition, the VL-domain was purified by another method that included sequential dialysis of the guanidine hydrochloride-soluble fraction of the inclusion bodies against 0.1 M Tris-HCl buffers (pH 8.0) containing 5, 3, and 1 M urea. Contaminating proteins precipitated and were removed by centrifugation after each step of dialysis. Followed by refolding, this purification method yields the recombinant VL-domain with at least 90% purity (Fig. 3). Analysis of the antigen-binding activity of the VL-domain purified by the two described methods did not detect differences in affinity between the two types of preparations of the recombinant protein. Thus, protein purified by controlled dialysis was used in further functional studies of the VL-domain. IMAC was used to prepare highly purified protein for crystallization studies (manuscript in preparation).Fig. 3. Purification of the VL-domain without IMAC: 1, 2, 3) soluble fraction of inclusion bodies after dialysis against buffers containing 5, 3, and 1 M urea, respectively; 4) isolated VL-domain after refolding; 5) refolded VL-domain purified by IMAC; M, molecular weight markers including bovine serum albumin (66 kD), ovalbumin (45 kD), glyceraldehyde-3-phosphate dehydrogenase (36 kD), carbonic anhydrase (29 kD), chymotrypsinogen (24 kD), trypsin inhibitor (20 kD), and alpha-lactalbumin (14 kD).
Refolding of the recombinant VL-domain was performed by controlled dialysis, substituting urea-free buffer for the buffer containing 1 M urea (10% buffer volume per h). This dialysis protocol facilitates the use of relatively high protein concentrations (in this case, 0.4-0.8 mg/ml), whereas in most studies of recombinant fragments of antibodies the concentrations during refolding did not exceed 0.1 mg/ml. Moreover, the controlled dialysis method increases the refolding yield up to 100% depending on the initial concentration of the samples. The concentration of 0.4 mg/ml provides for almost complete recovery of the VL-domain during refolding. Two-fold increase in the recombinant protein concentration decreases the refolding yield to 75%, but this number significantly exceeds the yields described by other authors varying from 10 to 60% [7, 25]. In several studies, the refolding yield reached 100% but the protein concentrations did not exceed 30 µg/ml [26]. Increasing the rate of dialysis during refolding, according to our data, decreases the yield of the soluble protein.
Thus, the expression, purification, and refolding protocols yield soluble recombinant VL-domain with high concentration and at least 95% purity according to SDS-polyacrylamide gel electrophoresis (Fig. 3).
Analysis of the antigen-binding activity of the recombinant VL-domain. Antigen binding by the VL-domain was analyzed by two variants of ELISA as described in “Materials and Methods”. Both ELISA systems detected functional activity of the recombinant protein, but the parameters of interaction of the VL-domain with ferritin significantly varied between two ELISA schemes. This is due to the presence of various forms of ferritin in the analytical systems.
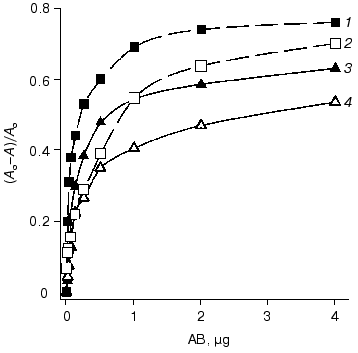
The data of competitive ELISA with immobilized antigen indicate that the VL-domain interacts with the antigen with association constant (1.8-2.1)·107 M-1, which is 30-fold lower than that of the parental antibody F11. It should be noted that the association constant of the VL-domain with ferritin did not depend on the method of antigen immobilization (Fig. 4). In contrast, the affinity of antibody F11 was 4-6-fold higher when the antigen was immobilized via a spacer including polyclonal rabbit antibodies versus the affinity determined in the case of ferritin directly immobilized on the polystyrene surface.
The association constant of the polystyrene-absorbed VL-domain with ferritin was higher in noncompetitive ELISA than that in competitive ELISA (Fig. 5). Immobilization of the VL-domain on the surface of polystyrene can facilitate the formation of the antigen-binding site of the recombinant antibody. The association constant of monoclonal antibody F11 and ferritin in noncompetitive ELISA is 6.5·108 M-1 (Fig. 5). Thus, the affinity of the parental antibody versus that of the isolated recombinant VL-domain in this system differed by only 5-8-fold, suggesting that the antigen-binding activity of the recombinant protein is similar to that of the authentic immunoglobulin.Fig. 4. Competitive ELISA of ferritin binding by isolated VL-domain (solid line) and full-length monoclonal antibody F11 (dotted line). Increasing amounts of the antibodies competed with the F11-biotin conjugate for binding to absorbed ferritin. A and A0, activity of peroxidase bound in the presence and in the absence of the competing agent, respectively; AB, amount of competing antibodies in the sample. Ferritin was immobilized on the polystyrene surface either directly (2, 4) or via a spacer of immobilized polyclonal rabbit antibodies (1, 3). The association constants were determined by the double reciprocal method; the values for the VL domain were 1.9·107 (3) and 2.0·107 M-1 (4) and the values for antibody F11 were 5.4·108 (1) and 1.1·108 M-1 (2).
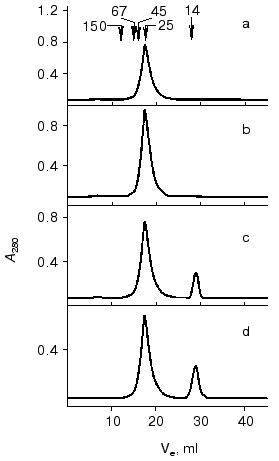
Gel-filtration. To determine the molecular forms of the soluble VL-domain after purification and refolding, the preparation was subjected to gel-filtration. Variation of the recombinant protein concentration from 1 to 10 mg/ml did not influence the elution pattern (Fig. 6, a and b); the VL-domain migrated almost exclusively as a dimer with 25-27-kD apparent molecular weight (the calculated weight of the dimer is 26.3 kD). Only when the protein concentration was decreased to 0.5 mg/ml, a peak with ~13.5-kD apparent molecular weight appeared, corresponding to the monomeric form of the recombinant protein (Fig. 6c). However, the fraction of monomer calculated from the peak area was low, not exceeding 10%. Further decrease in protein concentration below 50 µg/ml still resulted in significant dimerization of the recombinant fragment, and the fraction of the monomeric form of the VL-domain slightly increased, to 15-20% of the total protein (Fig. 6d).Fig. 5. Noncompetitive ELISA of ferritin binding by antibody F11 (b) and its isolated VL-domain (a); Ka = 6.5·108 (b) and 1.2·108 M-1 (a), respectively. Immobilized VL-domain or antibody F11 were interacted with increasing amounts of ferritin.
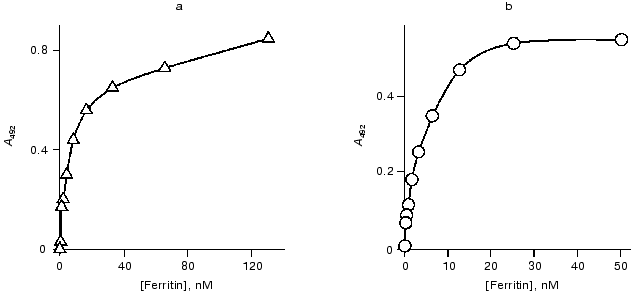
Fig. 6. FPLC of VL-domain preparations through a Superose 12, HR 10/3 column (1.5 × 35 cm); Ve corresponds to the elution volume. Protein concentration was 10 (a), 1 (b), 0.5 (c), and 0.05 mg/ml (d). Arrows correspond to the elution volumes of the standard proteins including immunoglobulin G (150 kD), bovine serum albumin (67 kD), ovalbumin (45 kD), chymotrypsinogen (25 kD), and alpha-lactalbumin (14 kD).
DISCUSSION
The present work demonstrates that the bacterially expressed, purified, and refolded VL-domain preserves its antigen-binding activity. The constant of the VL-domain binding to ferritin is comparable to that of the full-length parental antibody. Data on the functional activity of isolated variable domains of immunoglobulins are scarce and contradictory. For example, the antigen-binding activity was not detected in the isolated recombinant VH- and VL-domains in one study [22]. On the other hand, isolated VH-domains were shown to interact with the antigens in others [4, 24, 27, 28]. The antigen-binding activity was detected in the case when the VL-domain is included in the light chain of an antibody comprising two domains, variable VL and constant CL [29]; the VL-domain activity was shown in a recombinant immunotoxin comprising the VL-domain of an antibody and a fragment of Pseudomonas exotoxin [8]. The latter work also reported that the VL-containing immunotoxin is more active than the construct comprising the same toxin fragment and the VH-domain. These data disagree with the accepted point of view that the impact of the VH-domain (included in authentic immunoglobulin) on the formation of the antigen-binding site is more significant than the impact of the VL-domain [7]. Our results are the first experimental evidence of the isolated recombinant VL-domain having antigen-binding activity.
The association constant of the VL-domain with ferritin was determined in competitive ELISA and was 30-fold lower than the affinity of the parental monoclonal antibody F11. This suggests the impact of the VH-domain on the formation of the antigen-binding site. Quantitative estimation of this impact is difficult without direct measurement of antigen-binding constant of VH-domain. Inability of the VL-domain to discriminate between the conformations of the antigen in the case of various protocols of its immobilization is another important result. The binding constant of the parental monoclonal antibody F11 and ferritin differed by ~5-fold between ELISA using two forms of the antigen, i.e., direct immobilization of ferritin on the polystyrene surface and immobilization using a spacer of rabbit polyclonal antibodies. In contrast, the association constant of the VL-domain with ferritin is similar for these two forms of the antigen. This indicates that conformational mobility of the antigen is important for increased affinity of the molecules participating in the immunochemical reactions. Moreover, the data suggest that the VL-domain (in the absence of VH-domain) forms a more open antigen-binding site with lower affinity and decreased number of conformational limitations. This facilitates conformational adjustment of the antigen-binding site during interaction of the antibody with various conformations of the antigen. More detailed analysis requires similar studies with the isolated VH-domain of the monoclonal antibody F11. Nevertheless, the data on the antigen-binding activity of the isolated VL-domain present new information on the mechanisms of the formation of the antigen-binding site of the immunoglobulin molecule, and this information can be of physiological importance.
Dimerization of the isolated recombinant VL-domain of an antibody was described previously [7, 23, 30] and possible mechanisms of dimer formation were suggested. The association constants of VL-domain homo-dimerization vary from 103 to 107 M-1 [31, 32]. Our soluble VL-domain exists as dimer at concentration 1-10 mg/ml. The small amount of the monomeric form appears only when the concentration was decreased to 0.5 mg/ml (~4·10-5 M), indicating that the dimerization of the VL-domain is concentration-dependent. It should be noted that the dimerization constants were calculated in experiments when the monomers were the minor components [31, 32]. Stability of the monomeric VL-domain is questionable, and dimerization may be a mechanism of stabilization of the VL-domain molecule, accounting for its solubility. Considering the data of the present work on the functional activity of the dimer, we suggest that dimerization of the isolated VL-domain may also be required for the formation of an alternative antigen-binding site. This site of the homodimer has lower affinity than that of the authentic site of the VL-VH heterodimer, but it has enhanced steric adaptation to the conformational forms of the antigen (possibly due to increased conformational mobility of the alternative site). However, direct comparison of the antigen-binding affinity of the monomeric form of the VL-domain with that of the VL homodimer is required to confirm this hypothesis.
The work was supported by the Belarus Foundation for Basic Research (grant No. B98-221) and the European Molecular Biology Organization (grant No. ASTF-8609).
REFERENCES
1.Bird, R. E., Hardman, K. D., Jacobson, J. W.,
Johnson, S., Kaufman, B. M., Lee, S.-M., Lee, T., Pope, S. H., Riordan,
G. S., and Whitlow, M. (1988) Science, 242, 423-426.
2.Huston, J. S., Levinson, D., Mudget-Hunter, M.,
Tai, M.-S., Novotny, J., Margolies, M., Ridge, R. J., Bruccoleri, R.
E., Haber, E., Crea, R., and Oppermann, H. (1988) Proc. Natl. Acad.
Sci. USA, 85, 5879-5893.
3.Johnson, K. S., and Chiswell, D. J. (1993) Curr.
Opin. Struct. Biol., 3, 564-571.
4.Davies, J., and Riechmann, L. (1995)
Biotechnology (N. Y.), 13, 475-479.
5.Sheriff, S., and Constantine, K. L. (1996)
Nature Struct. Biol., 3, 733-736.
6.Brinkmann, U. (1996) Mol. Med. Today,
2, 439-446.
7.Givol, D. (1991) Mol. Immunol., 28,
1379-1386.
8.Brinkmann, U., Lee, B. K., and Pastan, I. (1993)
J. Immunol., 150, 2774-2782.
9.Hurle, M. R., Helms, L., Li, L., and Wetzel, R.
(1994) Proc. Natl. Acad. Sci. USA, 91, 5446-5450.
10.Stevens, F. J., Myatt, E. A., Chang, C.-H.,
Westholm, F. A., Eulitz, M., Weiss, D. T., Murphy, C., Solomon, A., and
Schiefer, M. (1995) Biochemistry, 34, 10697-10702.
11.Helms, L., and Wetzel, R. (1995) Prot.
Sci., 4, 2073-2081.
12.Martsev, S. P., Kravchuk, Z. I., Chumanevich, A.
A., Vlasov, A. P., Dubnovitsky, A. P., Bespalov, I. A., Arosio, P., and
Deyev, S. M. (1998) FEBS Lett., 441, 458-462.
13.Lunev, V. E., Mel'nikova, Ya. I., Koshkin, S. A.,
Luneva, N. M., Cherkesova, T. S., Vasilevskaya, I. A., Preigerzon, V.
A., and Martsev, S. P. (1993) Biochemistry (Moscow), 57,
745-758 (Russ.).
14.Martsev, S. P., Kravchuk, Z. I., Vlasov, A. P.,
and Lyakhnovich, G. V. (1995) FEBS Lett., 361,
173-175.
15.Kravchuk, Z. I., Chumanevich, A. A., Vlasov, A.
P., and Martsev, S. P. (1998) J. Immunol. Meth., 217,
131-141.
16.Bespalov, I. A., Shiyanov, P. A., and
Lukashevich, L. V. (1993) Mol. Biol. (Moscow), 27,
451-460.
17.Maniatis, T., Fritisch, E. F., and Sambrok, J.
(1982) in Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, N. Y.
18.Gill, S. C., and von Hippel, P. H. (1989)
Anal. Biochem., 182, 319-326.
19.Laemmli, U. K. (1970) Nature, 227,
680-685.
20.Van Dyke, M. W., Sirito, M., and Sawadogo, M.
(1992) Gene, 111, 99-104.
21.Dubel, S., Breitling, F., Klewinghaus, I., and
Little, M. (1992) Cell Biophys., 21, 69-79.
22.Worn, A., and Pluckhthun, A. (1998)
Biochemistry, 37, 13120-13127.
23.Raffen, R., Stewens, P. W., Boogaard, C.,
Schiffer, M., and Stevens, F. J. (1998) Prot. Eng., 11,
303-309.
24.Ward, E. S., Gussov, D., Griffiths, A. D., Jones,
P. T., and Winter, G. (1989) Nature, 341, 544-546.
25.Buchner, J., and Rudolph, R. (1991)
Bio/Technol., 9, 157-162.
26.Buchner, J., Pastan, I., and Brinkmann, U. (1992)
Anal. Biochem., 205, 263-270.
27.Chen, Y., and Stollar, B. D. (1999) J.
Immunol., 162, 4663-4670.
28.Reiter, Y., Schuck, P., Boyd, L. F., and Plaksin,
D. (1999) J. Mol. Biol., 290, 685-698.
29.Sun, M., Li, L., Gao, Q. S., and Paul, S. (1994)
J. Biol. Chem., 269, 734-738.
30.Stevens, F. J., Solomon, A., and Shiffer, M.
(1991) Biochemistry, 30, 6803-6805.
31.Stevens, F. J., Westholm, F. A., Solomon, A., and
Shiffer, M. (1980) Proc. Natl. Acad. Sci. USA, 77,
1144-1148.
32.Stevens, P. W., Ruffen, R., Hanson, D. K., Deng,
Y.-L., Hammond, M. B., Westholm, F. A., Murphy, C., Eulitz, M., Wetzel,
R., Solomon, A., Schiffer, M., and Stevens, F. J. (1995) Prot.
Sci., 4, 421-432.