A Study of the Asp110-Glu112 Region of EcoRII Restriction Endonuclease by Site-Directed Mutagenesis
V. N. Sergeev, S. E. Chalov, V. L. Drutsa, and E. S. Gromova*
School of Chemistry and Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: gromova@biorg.chem.msu.su* To whom correspondence should be addressed.
Received November 18, 1999; Revision received June 19, 2000
Site-directed mutagenesis of the ecoRII gene has been used to search for the active site of the EcoRII restriction endonuclease. Plasmids with point mutations in ecoRII gene resulting in substitutions of amino acid residues in the Asp110-Glu112 region of the EcoRII endonuclease (Asp110 --> Lys, Asn, Thr, Val, or Ile; Pro111 --> Arg, His, Ala, or Leu; Glu112 --> Lys, Gln, or Asp) have been constructed. When expressed in E. coli, all these plasmids displayed EcoRII endonuclease activity. We also constructed a plasmid containing a mutant ecoRII gene with deletion of the sequence coding the Gln109-Pro111 region of the protein. This mutant protein had no EcoRII endonuclease activity. The data suggest that Asp110, Pro111, and Glu112 residues do not participate in the formation of the EcoRII active site. However, this region seems to be relevant for the formation of the tertiary structure of the EcoRII endonuclease.
KEY WORDS: site-directed mutagenesis, ecoRII gene, EcoRII restriction endonuclease mutants
Type II restriction endonucleases are enzymes that interact specifically with DNA; in the presence of Mg2+, they cleave the DNA in certain sites. More than 2900 such enzymes are known at present [1], many of them being widely used in genetic engineering. However, the mechanism of their interaction with substrate DNAs remains insufficiently studied. Amino acid sequences of many restriction enzymes have been determined; as a rule, the enzymes display no marked homology in their primary structures [2, 3]. Data on the crystal structures of five type II restriction endonucleases (EcoRI, EcoRV, BamHI, PvuII, and Cfr10I) as well as their DNA complexes are now available [2, 3]. From X-ray structure analysis, the amino acids participating in the formation of the active sites of these enzymes have been determined, and a similarity in their active site structures is revealed. A basic motif for the amino acid sequences in the active sites was proposed: P (E/D) Xn (E/D) Z K. This motif is not completely conservative. For instance, the proline residue may be absent in some restriction endonuclease active sites [2-4]. Also, it is known that both the FokI (a type IIs restriction endonuclease whose crystal structure was recently determined) [5, 6] and the phage lambda exonuclease [7] active sites resemble the active sites of the five above-mentioned restriction endonucleases. These data suggest structural similarity of restriction endonuclease active sites [2-4, 8].
EcoRII restriction endonuclease belongs to the type IIe restriction enzymes, which need the recognition of two DNA sites for their catalytic act [9]. The active enzyme-substrate complex consists of two identical subunits of EcoRII endonuclease interacting with two specific DNA sites [10]. But the crystal structure of EcoRII restriction endonuclease as well as of other IIå type enzymes has not been determined. Homology exists between the C-terminal amino acid sequence of EcoRII endonuclease and motifs characterizing the integrase family of recombinases [11]. Recently, the sites responsible for specific DNA-binding were determined in EcoRII endonuclease [12]. Of special interest is the determination of amino acid residues involved in the active site of EcoRII endonuclease. Site-directed mutagenesis is one of the most effective approaches in such investigations. In the present study, we attempted to use this method for evaluation of the involvement of distinct amino acid residues in formation of the active site of EcoRII restriction endonuclease. To do this, we selected a region in the EcoRII endonuclease molecule for “protein design”, performed site-directed mutagenesis to produce a number of mutant ecoRII genes, expressed these genes, and then tested the gene products to determine whether they possessed EcoRII endonuclease activity.
MATERIALS AND METHODS
The enzymes T4 polynucleotide kinase, T4 DNA ligase, T4 DNA polymerase, E. coli DNA polymerase I, exonuclease III, and BamHI and HindIII restriction endonucleases were purchased from MBI Fermentas (Lithuania), EcoRII restriction endonuclease was provided by N. I. Matvienko (Institute of Protein Research, Russian Academy of Sciences, Pushchino, Moscow Region, Russia).
The JM109 strain of E. coli (Stratagene, USA) was used for gene engineering work. The pR224 plasmid containing the ecoRII gene was kindly provided by Professor A. Bhagwat (Wayne State University, Detroit, USA).
Oligodeoxyribonucleotides were synthesized using a standard automated
solid-phase amidophosphite method. We used the individual
oligodeoxyribonucleotides:
d(CCCCCTTGCTCATCA) (I)
and
d(pCTGACGCTCCTGGCTTTCA) (II)
as well as mixtures of compounds of similar structure:
d(pGCCCACTTCAGVHWCCTGAAAATACAGGGGCT) (III),
d(pCCACTTCAGGATSNTGAAAATACAGGGGCT) (IV),
and
d(pCTTCAGGATCCTVHWAATACAGGGGCT) (V),
where N = A, C, G, or T; V = A, C, or G; H = A, C, or T;
W = A or T; S = C or G.
The mutagenic oligodeoxyribonucleotides III-V were synthesized using balanced mixtures of nucleotide synthons for appropriate positions [13].
Mutagenesis. The pR224 plasmid has two HindIII restriction sites. To remove one that lies beyond the boundaries of the ecoRII gene, we treated the pR224 plasmid with HindIII endonuclease (18°C, 2 min), isolated the linearized plasmid (which was cleaved at only one of two HindIII restriction sites) by agarose gel electrophoresis, processed the cohesive ends of the plasmid with T4 DNA polymerase, and ligated them with DNA ligase. The resulting product was used for transformation of E. coli cells, and cloned plasmids were selected by restriction analysis. The pR224H plasmid contained a single HindIII restriction site inside the ecoRII gene. The nucleotide sequence of the ecoRII gene matched the previously reported sequence [14] with the exception of the region encoding amino acid residues 103-113, this being in agreement with [15].
Site-directed mutagenesis of the “Asp110” and “Pro111” codons of the ecoRII gene was performed using the pR224H plasmid. To facilitate initial selection of mutants, site-directed mutagenesis of the “Glu112” codon of the ecoRII gene was performed using the previously constructed mutant plasmid pR224HB (“Pro111” --> “Ala111”), which had no BamHI restriction site.
Nucleotide substitutions in the ecoRII gene were performed as described in [16] using plasmids purified by centrifugation in a CsCl density gradient [17]. We cleaved plasmids pR224H or pR224HB with HindIII restriction endonuclease, partially hydrolyzed them with exonuclease III to obtain single-stranded ends of length 100-200 nucleotides, hybridized with both 15-20% molar excess of the mutagen oligodeoxyribonucleotide (III, IV, or V) and a pair of adapter oligodeoxyribonucleotides (I and II), then processed with DNA ligase to obtain a circular form, built-up the single-stranded sequences with T4 DNA polymerase, and processed again with DNA ligase. The initial stages of DNA restructuring, namely, the cleavage at the HindIII site and the partial hydrolysis with exonuclease III, were performed with monitoring using 0.6% agarose gel electrophoresis. The resulting heteroduplex was sedimented with ethanol, treated with DNA polymerase I, and then used for the transformation of E. coli cells. We examined the existence of HindIII and BamHI restriction sites in cloned plasmids. The HindIII restriction site should persist in all “target” plasmid mutations, that is, in which the nucleotide sequence of the mutating region corresponds to the sequence of the mutagenic oligodeoxyribonucleotides used. The BamHI restriction site should disappear in mutants in both the “Asp110” and “Pro111” codons, whereas it should be restored in mutants in the “Glu112” codon. The nucleotide sequence of the region subjected to site-directed mutagenesis was examined in previously selected plasmids using Sanger's sequencing method.
To obtain a deletion mutant, the pR224H plasmid was cleaved at the unique BamHI restriction site, the linearized plasmid was treated with nuclease S1 for removing the single-stranded and small double-stranded terminal sequences, and then ligated with DNA ligase. After the transformation of E. coli cells, the clones expressing the mutant EcoRII restriction endonuclease forms were screened using a direct electrophoretic assay (10% SDS-PAGE) of cell-extract proteins. The nucleotide sequence of the deletion region of the ecoRII gene was determined in plasmids extracted from the cells expressing EcoRII proteins using Sanger's sequencing method.
All gene engineering work was performed using standard protocols [17].
Revealing of the mutant forms of EcoRII enzyme and determination of EcoRII endonuclease activity. Cell extracts were prepared as described previously [14] with slight modifications. E. coli cells from 0.5 ml of overnight culture were harvested by centrifugation, suspended in 50 µl of 0.05 M sodium phosphate, pH 8.8, containing 0.3 M NaCl and lysozyme (4 mg/ml), incubated 15 min on ice, and disrupted by two freeze-thaw treatments. The resulting suspension was centrifuged (15 min, 12,000g, 4°C), and the supernatant (“cell extract”) was mixed with two volumes of ice-cold buffer for storage of the EcoRII endonuclease (0.2 M Tris-HCl, pH 7.5, 0.25 M NaCl, 0.025 M MgCl2, 0.035 M dithiothreitol, 50% glycerol) and used directly for the measurement of EcoRII endonuclease activity (see below). Pellets were washed with 0.2 ml of 0.05 M sodium phosphate buffer (pH 8.8) containing 0.3 M NaCl and Triton X-100 (1%), then washed with the same buffer without Triton X-100, dissolved in 60 µl of 0.062 M Tris-HCl (pH 6.8) containing 2-mercaptoethanol (5%), SDS (2%), and urea (8 M). The solution was heated for 5 min at 90°C, and the proteins were assayed by electrophoresis in 10% polyacrylamide gel with SDS.
For determination of EcoRII endonuclease activity, non-methylated pBR322 plasmid (0.3 µg) was added to the cell extracts (2 µl) in 0.05 M Tris-HCl buffer (pH 7.5) containing 0.1 M NaCl and 0.01 M MgCl2 (10 µl total volume). The mixture was incubated for 40 min at 37°C and examined by electrophoresis in 1.4% agarose gel.
All tests and assays were repeated at last twice for all plasmid constructions described, producing identical results.
RESULTS AND DISCUSSION
We chosen a region of the EcoRII restriction endonuclease for mutagenesis taking into consideration the crystal structure of EcoRI, EcoRV, BamHI, PvuII, and Cfr10I restriction endonucleases as well as the general structure of their active sites--the P (E/D) Xn (E/D) Z K motif [2-4]. The EcoRII restriction endonuclease has at least three such motifs: P76 D77 X18 E96 K97, D110 P111 E113 X17 D130 C131 K132, and P314 V315 E316 X22 D339 K340. The region D110-P111-E112 in motif D110 P111 E113 X17 D130 C131 K132 was chosen as the first to be subjected to mutation analysis. We planned to substitute the amino acid residues in each position specified. The substitution of Asp110, Pro111, and/or Glu112 should result in complete loss of enzymatic activity if each of these residues or at least one of them is involved in formation of the EcoRII restriction endonuclease active site, as has been demonstrated for the substitution of Asp74 or Asp90 in the EcoRV endonuclease molecule [18].
Oligodeoxyribonucleotides were designed for the site-directed mutagenesis so that we could obtain a full set of planned substitutions in the individual codon for the chosen amino acid residue after a single round of mutagenesis. Three mixtures of mutagenic oligodeoxyribonucleotides were used (III, IV, or V, see “Materials and Methods”), each resulting in the ecoRII gene mutation set characterized by substitution of Asp110, Pro111, or Glu112, respectively, in the protein molecule.
Site-directed mutagenesis was performed in the following region of the
ecoRII gene (the nucleotide substitution area is marked by the
bold symbols):
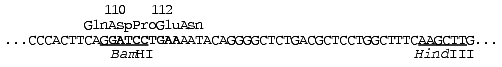
Initial selection of target mutant clones was performed using plasmid hydrolysis with both HindIII and BamHI endonucleases. Restriction analysis displayed a mutagenesis pattern characteristic for the method used [16]. A high yield (>96%) of plasmids with restored HindIII site was achieved in all experiments, indicating accurate following of the steps of the initial plasmid treatment protocol. Most of the plasmids analyzed after mutagenesis (70-76%) were identical to the initial plasmid. Approximately 21-24% of the plasmids (target mutants) demonstrated some modifications in the region of the BamHI site. Three to six percent of the plasmids (side mutations, mainly deletions) demonstrated lack of the HindIII site and alterations (in some instances significant decrease) in their molecular masses. The average yield of target mutants was calculated as the ratio of the number of BamHI endonuclease-resistant clones (disappearance of the BamHI site in the case of mutagenesis in the region of the “Asp110” or “Pro111” codons) or BamHI endonuclease-sensitive clones (restoration of BamHI site in the case of mutagenesis in the region of the “Glu112” codon) to the total number of clones analyzed. Yields of target mutants were 22% (12 out of 54 clones) for mutagenesis in the region of the “Asp110” codon, 24% (10 out of 42 clones) for mutagenesis in the region of the “Pro111” codon, and 21% (10 out of 48 clones) for mutagenesis in the region of the “Glu112” codon. Sequencing revealed the following codon substitutions: GAT (Asp110) --> AAA, AAT, ACT, GTA, or ATT; CCT (Pro111) --> CGT, CAT, GCT, or CTT; GAA (Glu112) --> AAA, CAA, or GAT (see table).
Mutant EcoRII proteins with substituted or deleted amino acid
residues
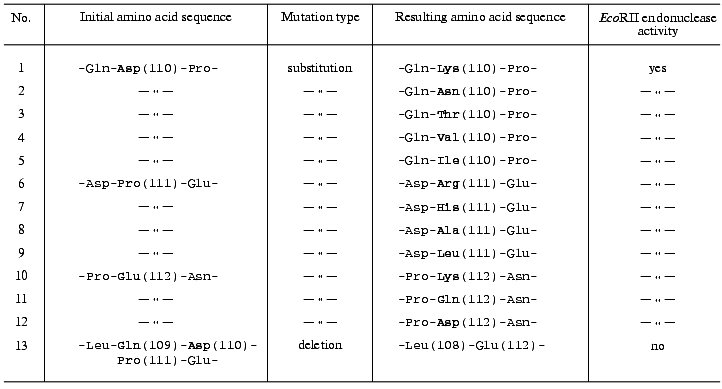
Aside from above-listed plasmids, which have codon substitutions, the plasmid carrying deletion of the sequence CAGGATCCT encoding amino acids Gln109-Pro111 in the ecoRII gene (table) was designed using standard gene engineering methods (plasmid cleavage at the unique BamHI site, hydrolysis with S1 nuclease, ligation) with the screening of clones expressing the “full-size” ecoRII gene product.
Direct assay of cell proteins in SDS polyacrylamide gels by the method of Laemmli indicated that E. coli JM109 strains carrying all modified plasmids produce mutant proteins with the same electrophoretic mobility as the EcoRII endonuclease from E. coli JM109 carrying the pR224H plasmid. Tests on the ability to specifically cleave a standard DNA revealed EcoRII endonuclease activity in all cell extracts of E. coli JM109 strains carrying all substitution variants for all amino acid residue codons (table). In contrast, there was no EcoRII endonuclease activity in the cell extract from the E. coli JM109 strain carrying a plasmid with the deletion in the ecoRII gene.
Thus, 12 mutants resulting from site-directed mutagenesis have been characterized that have codon substitutions in a distinct site of the ecoRII gene. The expression of the mutant genes in E. coli cells resulted in mutant EcoRII proteins with point amino acid substitutions: Asp(110) --> Lys, Asn, Thr, Val, or Ile; Pro(111) --> Arg, His, Ala, or Leu; Glu(112) --> Lys, Gln, or Asp. All these proteins demonstrated EcoRII endonuclease activity. We can exclude the accidental preservation of catalytic activity because the amino acid residues in each position were replaced by different ones possessing different and in some cases opposite properties. Thus, it is reasonably safe to conclude that the Asp110-Glu112 region of the EcoRII enzyme does not belong to the catalytic site. Moreover, none of the residues Asp110, Pro111, and Glu112 is directly involved in formation of this site. However, the experiment on the deletion of the codons for amino acid residues 109-111, resulting in complete loss of EcoRII endonuclease activity, suggests that this region plays an important role in formation and/or maintenance of the tertiary structure of the EcoRII restriction endonuclease.
Thus, the question of which amino acid residues are involved in formation of the EcoRII restriction endonuclease active site remains unsolved. However, it was recently reported that mutant E96Aof EcoRII endonuclease (mutation in the region of the P76 D77 X18 E96 K97 motif) lacks the ability to bind and cleave DNA [12]. This report and ours narrow the boundaries of where the active site may be found and allow us to plan further studies on the mutagenesis of EcoRII. These studies are currently in progress in our laboratory.
We are grateful to professor A. Bhagwat (Wayne State University, Detroit, MI, USA) for providing us with the pR224 plasmid. This study was supported by the Howard Hughes Medical Institute (grant HHMI 75195-545501), the Russian State Scientific Program “Newest Methods in Bioengineering”, direction “Protein Engineering” (project No. 3-07), and the Russian Foundation for Basic Research (grant No. 98-04-49108).
REFERENCES
1.Roberts, R. J., and Macelis, D. (1998)
Nucleic Acids Res., 26, 338-350.
2.Aggarwal, A. K. (1995) Curr. Opin. Struct.
Biol., 5, 11-19.
3.Pingoud, A., and Jeltsch, A. (1997) Eur. J.
Biochem., 246, 1-22.
4.Anderson, J. E. (1993) Curr. Opin. Struct.
Biol., 3, 24-30.
5.Wah, D. A., Hirsch, J. A., Dorner, L. F.,
Schildkraut, I., and Aggarwal, A. K. (1997) Nature, 388,
97-100.
6.Wah, D. A., Bitinaite, J., Schildkraut, I., and
Aggarwal, A. K. (1998) Proc. Natl. Acad. Sci. USA, 95,
10564-10569.
7.Kovall, R. A., and Matthews, B. W. (1998) Proc.
Natl. Acad. Sci. USA, 95, 7893-7897.
8.Skirgaila, R., Grazulis, S., Bozic, D., Huber, R.,
and Siksnys, V. (1998) J. Mol. Biol., 279, 473-481.
9.Krueger, D. H., Kupper, D., Meisel, A., Reuter, M.,
and Schroeder, C. (1995) FEMS Microbiol. Rev., 17,
177-184.
10.Petrauskene, O. V., Karpova, E. A., Gromova, E.
S., and Guschlbauer, W. (1994) Biochem. Biophys. Res. Commun.,
198, 885-890.
11.Topal, M. D., and Conrad, M. (1993) Nucleic
Acids Res., 21, 2599-2603.
12.Reuter, M., Schneider-Mergener, J., Kupper, D.,
Meisel, A., Mackeldanz, P., Krueger, D. H., and Schroeder, C. (1999)
J. Biol. Chem., 274, 5213-5221.
13.Drutsa, V. L., Krivonos, A. V., Koroleva, O. N.,
and Shabarova, Z. A. (1990) Bioorg. Khim., 16,
1052-1059.
14.Bhagwat, A. S., Johnson, B., Weule, K., and
Roberts, R. J. (1990) J. Biol. Chem., 265, 767-773.
15.Kossykh, V., Repyk, A., Kaliman, A., and
Buryanov, Y. (1989) Biochim. Biophys. Acta, 1009,
290-292.
16.Drutsa, V. L., and Kaberdin, V. R. (1992)
Nucleic Acids Res., 20, 922.
17.Maniatis, T., Fritsch, E. F., and Sambrook, J.
(1981) Molecular Cloning, Cold Spring Harbor, N. Y.
18.Selent, U., Rueter, Th., Koehler, E., Liedtke,
M., Thielking, V., Alves, J., Oelgeschlaeger, Th., Wolfes, H., Peters,
F., and Pingoud, A. (1992) Biochemistry, 31,
4808-4815.