REVIEW: Interactions between Carnosine and Zinc and Copper: Implications for Neuromodulation and Neuroprotection
P. Q. Trombley*, M. S. Horning, and L. J. Blakemore
Biomedical Research Facility, Department of Biological Science, Florida State University, Tallahassee, Florida 32306-4340, USA; fax: (850) 644-0989; E-mail: trombley@neuro.fsu.edu* To whom correspondence should be addressed.
Received November 22, 1999
This review examines interactions in the mammalian central nervous system (CNS) between carnosine and the endogenous transition metals zinc and copper. Although the relationship between these substances may be applicable to other brain regions, the focus is on the olfactory system where these substances may have special significance. Carnosine is not only highly concentrated in the olfactory system, but it is also contained in neurons (in contrast to glia cells in most of the brain) and has many features of a neurotransmitter. Whereas the function of carnosine in the CNS is not well understood, we review evidence that suggests that it may act as both a neuromodulator and a neuroprotective agent. Although zinc and/or copper are found in many neuronal pathways in the brain, the concentrations of zinc and copper in the olfactory bulb (the target of afferent input from sensory neurons in the nose) are among the highest in the CNS. Included in the multitude of physiological roles that zinc and copper play in the CNS is modulation of neuronal excitability. However, zinc and copper also have been implicated in a variety of neurologic conditions including Alzheimer's disease, Parkinson's disease, stroke, and seizures. Here we review the modulatory effects that carnosine can have on zinc and copper's abilities to influence neuronal excitability and to exert neurotoxic effects in the olfactory system. Other aspects of carnosine in the CNS are reviewed elsewhere in this issue.
KEY WORDS: carnosine, zinc, copper, glutamate receptors, synaptic transmission, tissue culture, electrophysiology, neuromodulation, neurotoxicity, olfactory system, olfactory bulb, rat, Alzheimer's disease, Parkinson's disease
CARNOSINE IN THE CENTRAL NERVOUS SYSTEM
Carnosine (beta-alanyl-L-histidine) is a dipeptide that is expressed in glial cells throughout the brain as well as in neuronal pathways of the visual and olfactory systems [1-3]. Throughout the brain, there is an almost complete overlap of glial fibrillary acidic protein and carnosine immunoreactivity, and carnosine can be synthesized by rodent glial cells in culture [4, 5]. Therefore, most carnosine-reactive cells are thought to be astrocytes, oligodendrocytes, or ensheathing cells. In the olfactory system, carnosine immunoreactivity is seen in cells with astrocyte-like morphology throughout all layers of the rodent olfactory bulb in both neonates and adults [1]. Perhaps more significantly, carnosine is expressed in high concentrations in the olfactory sensory neurons, as well as their terminals in the glomerular layer of the olfactory bulb [6]. Carnosine also meets many of the following criteria for a neurotransmitter.
1. Presence of the compound in presynaptic terminals. The concentration of carnosine in the olfactory bulb is high (2-5 mM) [7]. In addition to glial cells, it is present in presynaptic terminals and vesicles of olfactory sensory neurons, the source of afferent input to the olfactory bulb [3, 6, 8, 9].
2. Presence of enzymes for synthesis and degradation. Carnosine, its synthetic enzyme, carnosine synthetase, and its degradative enzyme, carnosinase, are localized in the cytosol of cells in the mouse olfactory bulb and epithelium [10]. In this study, most of the carnosine was found in synaptosomes isolated by sucrose-gradient centrifugation.
3. High-affinity uptake of the compound or its precursors. Burd and colleagues [11] reported that the majority of beta-[3H]alanine, administered to the nasal cavity of hamsters, was converted into carnosine that appeared in the olfactory nerve and glomerular layers of the olfactory bulb. In a separate study, it was reported carnosine could be sequestered by astroglia-rich primary cultures derived from brains of newborn mice [12]. Uptake and release of carnosine by these cells were found to be mediated by a saturable, high-affinity transport system with Km = 50 µM. More recently, a peptide/histidine transporter (PHT1) that can transport carnosine has been identified in both neuronal and nonneuronal cells in the rat [13].
4. Selective release under depolarizing conditions. Potassium-induced depolarizations can induce the release of carnosine from mouse olfactory bulb synaptosomes in a calcium-dependent manner [14]. It also has been reported that carnosine release from cultured rat oligodendrocytes can be stimulated by glutamate receptor activation [15]. This release is apparently dependent on elevated intracellular Ca2+ concentrations. The fact that carnosine release was enhanced in the presence of zinc provides another link between zinc and carnosine.
5. Presence of receptor/binding sites. Binding sites for carnosine have been reported in membranes prepared from mouse olfactory bulbs. The binding of L-[3H]carnosine was saturable, reversible, and stereospecific, and had a Kd of about 770 nM [16, 17].
6. Postsynaptic application mimics the electrophysiological response to presynaptic stimulation. It has been reported that carnosine evokes inward currents in rat olfactory bulb neurons from cultured slices [18]. However, most investigators report little or no direct effects of carnosine on olfactory bulb neurons [19-22], and recent evidence indicates that glutamate is the olfactory sensory neuron transmitter [23-25].
In spite of the supportive evidence, the lack of a direct effect of carnosine on the afferent targets of carnosine-containing olfactory sensory neurons casts doubt on its status as a neurotransmitter. However, because carnosine is colocalized with glutamate and zinc in olfactory sensory neurons, it has been proposed that carnosine may modulate the actions of glutamate or zinc [3]. Data from our work suggest that carnosine may indirectly modulate neuronal excitability by influencing the actions of zinc and copper on amino acid receptors and synaptic transmission.
Data from a separate series of studies also suggest that carnosine can reduce or prevent the neurotoxic effects of these metals. These actions may be due to a combination of metal chelation and free radical scavenging [26-30].
ZINC AND COPPER IN THE CENTRAL NERVOUS SYSTEM
A number of techniques have been used to localize and quantify levels of transition metals (primarily zinc) in the CNS, including histochemistry, stable-isotope dilution, and retrograde/anterograde techniques (for review see [31]). The three main sources of zinc in the brain are: 1) a vesicular pool located in synaptic vesicles of nerve terminals; 2) a membrane-bound, metalloprotein, or protein-metal complex pool involved in metal and nonmetabolic reactions, and 3) an ionic pool of free or loosely bound ions in the cytoplasm [32]. Whereas vesicular and membrane-bound pools are thought to exist in the CNS, the identification of a number of zinc-binding proteins in the CNS [33-35] makes the existence of “free” zinc in the cellular milieu less likely [32]. However, there is evidence for an intracellular ionic pool [36, 37].
Chelatable or labile zinc is defined as zinc that can be abstracted from the cellular proteins to which it is bound and represents zinc available to potentially assume a neuromodulatory role. The remainder of zinc is “tightly” bound to proteins where it acts in a structural capacity or as a component of an enzyme catalytic site. A variety of histological techniques have been used to detect chelatable zinc, including the Timm-Danscher reaction [38-41]. Although the sources of chelatable zinc also include loosely bound zinc and ionic zinc, the primary source of chelatable zinc is thought to be vesicular compartments [40-42]. Reportedly, up to 15% of brain zinc is contained in synaptic vesicles [32], and particularly high concentrations of labile zinc are found in the vesicular compartments of the hippocampus [40, 41, 43] and olfactory bulb [8, 44]. In the hippocampus and olfactory bulb, vesicular zinc is thought to be localized to neurons using glutamate as a neurotransmitter [23, 44-46]. Zinc released from glutamatergic hippocampal neurons has been estimated to reach extracellular concentrations of 100 to 300 µM (e.g., [47]).
Although fewer studies of copper exist, it is likely that the brain harbors similar pools of copper and that similar synaptic mechanisms govern its release from synaptic vesicles. The identification of copper sources have been complicated by the fact that the Timm-Danscher reaction (used to identify chelatable zinc) does not distinguish between the two metals. It has been estimated that following depolarization, the concentration of copper in the synpatic cleft reaches at least 100 µM [48, 49].
These concentrations of zinc and copper are sufficient to have neuromodulatory effects [19, 50] and to be neurotoxic [30].
While significant levels of zinc and copper have been found in many brain regions including the hippocampus, locus ceruleus, hypothalamus, olfactory bulb, and cortex, the olfactory bulb contains one of the highest concentrations of zinc [51, 52] and the highest concentration of copper [52] in the CNS. From atomic absorption spectroscopy, it was reported that the bulb contains 969.7 ± 58.1 and 335.9 ± 33.0 nmoles per g dry weight of zinc and copper, respectively [52]. These numbers are difficult to appreciate out of context. It is perhaps more meaningful to recognize that zinc and copper are among the 20 most abundant elements in the brain. CNS concentrations of zinc and copper are quite high when contrasted with those of more familiar neuroactive substances. For example, the concentration of zinc in gray matter (a few hundred micromolar) is 10-100 times higher than of neurotransmitters such as acetylcholine or the monoamines, and 1000-10,000 times higher than those of most neuropeptides [53]. Whereas zinc is found throughout the olfactory bulb, it appears to be most highly concentrated in the glomerular and granule cell layers [8]. Although the precise location of copper in the olfactory bulb has yet to be determined, recent data [44] and our unpublished observations suggest that both zinc and copper are present in mitral/tufted cells.
RELATIONSHIP BETWEEN ZINC, COPPER, AND CARNOSINE ON NEURONAL
EXCITABILITY
Although the role(s) transition metals play in the brain, including the olfactory system, has yet to be fully clarified, some progress has been made. It is known that these transition metals are necessary for many normal functions in the nervous system, and that deficiencies result in poor growth and development of the CNS [54].
Synaptically released zinc or copper may play a significant role in the synaptic physiology of amino acid transmitter pathways. This hypothesis is based on several lines of evidence. Synaptic activity can release zinc from nerve terminals [47, 55], and it has been reported that endogenous zinc is released from neurons in sufficient quantities to modulate GABAergic synaptic transmission [56-58]. In addition, zinc has been found to modulate both excitatory and inhibitory synaptic transmission [19, 50, 59, 60]. This modulation involves zinc's antagonism of some forms of N-methyl-D-aspartate (NMDA) and GABA receptors [50, 61, 62].
Although it has been demonstrated that copper, like zinc, is localized to synaptic terminals [63] and can be released by depolarization [48, 49], little is known about the potential neuromodulatory actions of copper. Recent results suggest that copper can modulate amino acid receptors [50, 64, 65] and purine receptors (e.g., [66]). However, the effects of copper, at least on glycine receptors, purine receptors, and on synaptic transmission, appear to be distinct from the effects of zinc [19, 50, 66].
Carnosine may play an important role in modulating the effects of zinc and copper on neuronal excitability. Original interest in determining whether carnosine could evoke a membrane current from olfactory bulb neurons was based on several features of carnosine. Carnosine meets many of the criteria for a neurotransmitter, and it has been proposed to be the neurotransmitter at olfactory sensory neuron-olfactory bulb neuron synapses. Therefore, it was examined whether carnosine could evoke a membrane current from olfactory bulb neurons in primary culture [19]. (The olfactory bulb contains two primary classes of neurons, mitral/tufted (M/T) cells and inhibitory interneurons, periglomerular cells and granule cells. Both M/T cells and periglomerular cells receive direct input from carnosine-containing olfactory sensory neurons.) Carnosine (up to 1 mM) was applied under voltage-clamp recording conditions for 5-60 sec. Carnosine failed to evoke a membrane current in either M/T cells or interneurons.
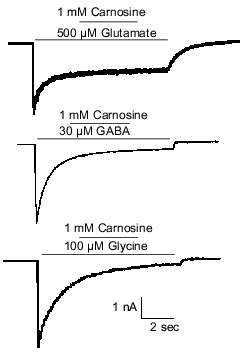
Because carnosine is colocalized with glutamate in olfactory sensory neurons [3, 23], it was hypothesized that carnosine may instead function as a neuromodulator of glutamate receptors. This hypothesis was tested by examining the effects of carnosine on glutamate-evoked currents in olfactory bulb neurons. Under voltage-clamp recording conditions, glutamate was applied at 500 µM to activate both the NMDA and AMPA/kainate subtypes of glutamate receptors. Co-application of 1 mM carnosine did not alter the glutamate-evoked currents in either M/T cells or interneurons (Fig. 1).
Olfactory glomeruli, the region of the olfactory bulb where olfactory sensory neurons make contact with olfactory bulb neurons, contain projections from interneurons that use GABA (e.g., [67]) and, perhaps, glycine as well [68, 69]. Therefore, it was investigated whether carnosine could modulate GABA or glycine receptors. As was the case for glutamate, coapplication of 1 mM carnosine failed to alter the current evoked by either GABA or glycine (Fig. 1). Thus, it was concluded that carnosine neither has a direct effect on olfactory bulb neurons nor directly alters currents mediated by glutamate, GABA, or glycine.Fig. 1. Carnosine does not directly modulate amino acid receptor-mediated currents from rat olfactory bulb neurons in primary culture. Under voltage-clamp recording conditions, application of 500 µM glutamate, 30 µM GABA, or 100 µM glycine evoked inward currents at a holding potential of -50 mV. A CsCl-based intracellular recording solution was used to produce a reversal potential near 0 mV for currents evoked by each amino acid. Carnosine (1 mM) had no effect when co-applied during the evoked currents.
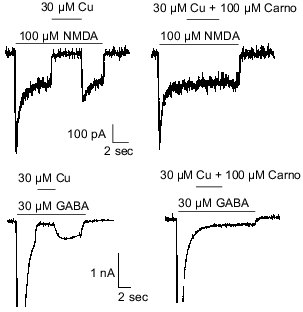
Carnosine is colocalized with zinc and/or copper in olfactory sensory neuron terminals [3, 8] and has been shown to chelate transition metal ions [27]. Therefore, it was hypothesized that carnosine may indirectly influence olfactory bulb neuron amino acid receptors by modulating the effects of zinc and/or copper previously observed at these sites [50]. The latter refers specifically to inhibition of NMDA- or GABA-evoked currents by zinc and copper. Therefore, carnosine was coapplied with zinc during currents evoked by NMDA or GABA in cultured olfactory bulb neurons. At concentrations up to 1 mM, carnosine only slightly reduced the effects of 30 µM zinc on NMDA- or GABA-receptor-mediated currents (Fig. 2). In contrast, 100 µM carnosine completely blocked the antagonistic action of 30 µM copper on membrane currents evoked by 100 µM NMDA or 30 µM GABA (Fig. 3). These findings support the hypothesis that carnosine can act as an indirect neuromodulator through modulation of the effects of zinc and copper.
Fig. 2. Carnosine (Carno) reduces the effects of zinc on GABA- and NMDA-evoked currents. Under voltage-clamp recording conditions, application of 30 µM zinc blocked most of the current evoked by 100 µM NMDA or 30 µM GABA. Carnosine (1 mM and 300 µM) reduced the antagonistic action of zinc when co-applied with zinc in case of NMDA- and GABA-evoked currents, respectively.
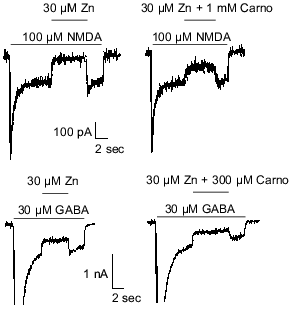
Fig. 3. Carnosine (Carno) blocks the effects of copper on GABA- and NMDA-evoked currents. Under voltage-clamp recording conditions, application of 30 µM copper blocked most of the current evoked by 100 µM NMDA or 30 µM GABA. Carnosine (100 µM) prevented the antagonistic action of copper when co-applied with copper in case of NMDA- and GABA-evoked currents.
EFFECTS OF ZINC, COPPER, AND CARNOSINE ON SYNAPTIC TRANSMISSION
Zinc and copper not only modulate amino acid receptors, but also can modulate excitatory and inhibitory synaptic transmission [19]. Figure 4 shows spontaneous excitatory synaptic activity of an olfactory bulb interneuron being driven by a glutamate-releasing M/T cell. Copper (30 µM) completely suppresses such activity [19]. Given the effects of carnosine on copper modulation of amino acid receptors, it was hypothesized that carnosine may similarly influence the actions of copper on synaptic transmission. Consistent with its effects on copper modulation of amino acid receptors, carnosine (300 µM) was found to completely prevent the actions of copper on synaptic transmission (Fig. 4). However, carnosine (100 µM-1 mM) alone had no direct effect on synaptic transmission. These results provided evidence to support the hypothesis that carnosine can indirectly influence synaptic transmission.
The effects of zinc (100 µM) on synaptic transmission are qualitatively similar to the effects of copper, but much less potent. The baseline potential is slightly hyperpolarized, and the frequency of the synaptic activity is reduced [19]. Given the findings about copper, it was hypothesized that carnosine also might influence the effects of zinc on synaptic transmission. Consistent with the slight effect of carnosine on zinc modulation of amino acid receptors, carnosine (1 mM) was found to only slightly affect the actions of 100 µM zinc on synaptic transmission (Fig. 4). As shown in Fig. 4, the effects of zinc, copper, and carnosine were rapid and easily reversible.Fig. 4. Carnosine modulates the actions of zinc and copper on synaptic transmission. Spontaneous excitatory synaptic activity, mediated by glutamate, was recorded in an olfactory bulb interneuron. Application of 30 µM copper dramatically reduced excitatory postsynaptic potentials and action potentials. Co-application of 300 µM carnosine completely prevented the antagonistic effects of 30 µM copper. The effects of 100 µM zinc on reducing excitatory synaptic transmission was less dramatic than the effects of copper. Co-application of 1 mM carnosine only slightly attenuated the effects of 100 µM zinc.
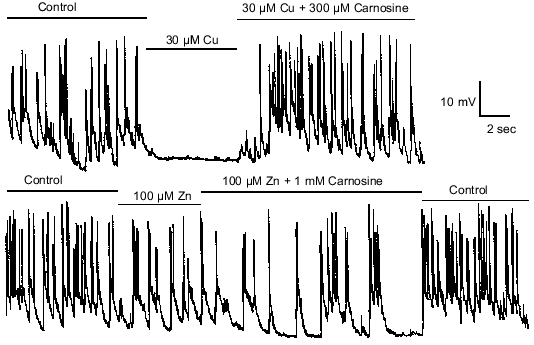
Zinc and copper also have been shown to suppress GABA-mediated spontaneous inhibitory synaptic transmission from interneurons onto olfactory bulb M/T cells. Therefore, it was investigated whether carnosine affects zinc and copper's effects on GABA-mediated synaptic transmission. The effects of carnosine on this zinc- and copper-mediated suppression of inhibitory transmission were found to parallel carnosine's effects on excitatory transmission; carnosine prevented the effects of copper, and reduced the effects of zinc.
RELATIONSHIP BETWEEN CARNOSINE AND ZINC AND COPPER ON NEURONAL
TOXICITY
Zinc imbalances due to shifts in brain zinc pools have increasingly been implicated in neurologic conditions, such as Alzheimer's disease, seizures, and ischemic stroke. Elevated or depressed levels of zinc in several brain regions have been documented in Alzheimer's disease patients by several investigators (for review, see [31]). In addition, translocation (influx) of zinc into postsynaptic limbic or cortical neurons has been observed following seizures induced by kainate [70]. Similar translocation of zinc released from presynaptic nerve terminals has been demonstrated in degenerating hippocampal hilar neurons following transient forebrain ischemia [71]. A combination of TSQ fluorescence and acid fuchsin staining was used to demonstrate that such zinc influx into hippocampal neurons precedes the neuronal degeneration induced by an ischemic insult [72]. Furthermore, it was demonstrated that such neuronal degeneration can be protected against by the application of Ca-EDTA (a metal chelator) prior to the transient ischemic insult [72]. In this study, Ca-EDTA also was found to protect cultured cortical neurons from the toxic effects of zinc, but not copper [72].
Toxic effects of zinc on hippocampal and cortical neurons also have been observed in models of Alzheimer's disease. Recent in vitro studies have shown that zinc induces beta-amyloid (betaA) protein clumping similar to that found in the senile plaques associated with Alzheimer's disease [73-77]. This aggregated (clumped) form of betaA protein has been found to be cytotoxic to hippocampal and cortical neurons, and it has induced apoptic cell death in in vitro studies [78]. betaA protein also has been shown to enhance the susceptibility of cultured human and murine cortical neurons to excitotoxic injury [79, 80]. Bush and colleagues have reported that low (i.e., physiological) concentrations of zinc (e.g., <5 µM) can aggregate betaA [73, 74].
Based in part on the above-described findings, Cuajungco and Lees [31] have proposed that rather than zinc initiating a neuropathological process de novo, cellular zinc levels may be perturbed to the point where zinc may contribute to disease progression (e.g., formation of plaques, triggering of inflammatory and/or degenerative responses). They identify three potential mechanisms by which zinc pools might be altered: 1) the release of excess cellular zinc (as seen with convulsive activity) [45]; 2) disruption of the blood brain barrier (as may occur with stroke) [81], and 3) oxidative stress (causing dissociation of zinc from metalloproteins or other proteins) [82-85]. It is also possible that changes in pH, as seen with seizure, ischemia, or inflammation can affect protein binding, further promoting dissociation of zinc (e.g., [86]).
A neuropathologic condition involving copper and oxidative damage is familial amyotrophic lateral sclerosis (FALS). A mutant form of copper-zinc superoxide dismutase (Cu,Zn-SOD), the enzyme critical for the detoxification of the superoxide radical produced during enzymatic oxidations, has been associated with this condition [87]. Other diseases with neuropathological components in which copper has been implicated include Alzheimer's disease, Menkes disease, Wilson's disease, and Pick's disease [88-91].
Olfactory dysfunction is a prominent feature in several neurological diseases in which zinc and copper have been implicated. For example, olfactory dysfunction and pathology is so evident in Alzheimer's disease that Ferreyra and Barragan [92] have proposed that Alzheimer's-associated pathology begins in olfactory regions. This is based on the finding that olfactory dysfunction is among the first symptoms of Alzheimer's disease, often involving a loss of olfactory perception and discrimination. Furthermore, the initial neuropathology associated with Alzheimer's disease--the formation of senile plaques and neurofibrillary tangles, the degeneration of neurons, a decrease in the density of dopamine receptors, and a decrease in tissue volume--occurs in the olfactory bulb [93, 94]. There also have been numerous conflicting reports regarding increased, normal, or decreased levels of zinc found in regions of the brain affected by Alzheimer's disease [31]. Of potential significance, recent studies documented significantly higher levels of zinc in the olfactory structures, hippocampus, and amygdala [95, 96]. Other inorganic ions that have shown marked variations in concentration with differing regions in the Alzheimer's diseased brain include copper, iron, aluminum, selenium, magnesium, silicon, and mercury [95, 97, 98].
Given the role of zinc and/or copper in many of these neuropathologies and the observed modulation of the effects of zinc and copper on synaptic transmission by carnosine, it was hypothesized that carnosine may protect against neurotoxicity mediated by these metals. Investigators first characterized the toxic effects of zinc and copper on olfactory bulb neurons in primary culture to provide a baseline [30].
Application of zinc or copper to cultured olfactory bulb neurons for 1 h was found to be associated with neuronal death [30]. Furthermore, death occurred at concentrations an order of magnitude below previously reported estimates of extracellular or synaptic concentrations of these transition metals (e.g., 30 µM)[47, 49]. Differences between the control group and all three of the experimental groups (10, 30, and 100 µM) were statistically significant (p < 0.001). The rate of cell death increased with increases in the concentration of zinc or copper to which cells were exposed. Even at the relatively low concentration of 30 µM, most neurons died within 5 h (Fig. 5).
Based on the report that chelation with EDTA reduces zinc-mediated toxicity [72] and the observed effects of carnosine on zinc/copper modulation of synaptic activity [19], it was hypothesized that endogenous carnosine also may protect againstmetal-induced toxicity via chelation.Fig. 5. Carnosine reduces the toxic effects of zinc and copper. a) In the experimental groups, 30 µM zinc or 30 µM zinc with 1 mM carnosine was applied for 1 h in the minimal essential medium (MEM) plus glucose (6 g/liter). After the 1 h application, the media containing zinc or zinc with carnosine were replaced with MEM plus glucose, and the extent of cell death was recorded every 30 min. Cell death was determined using Trypan blue and changes in morphology. Zinc (10-100 µM) was highly neurotoxic when applied for 1 h. Carnosine (1 mM) significantly reduced the toxic effects of 10 and 30 µM zinc, but not 100 µM zinc (only 30 µM zinc is shown). b) The rate of cell death in the presence of 30 µM copper or 30 µM copper plus 1 mM carnosine. The experimental protocol was the same as that for zinc. Carnosine significantly reduced the toxic effects of copper. Each line in both panels represents data from hundreds of neurons.
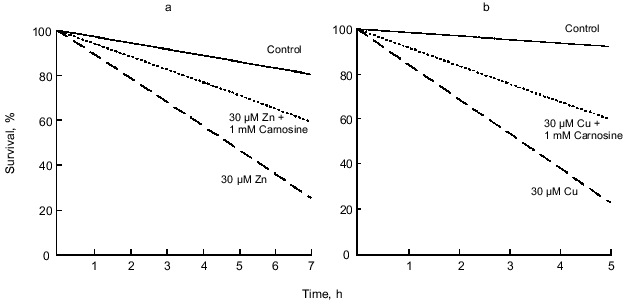
Carnosine and zinc or copper were coapplied to cultured olfactory bulb neurons for 1 h. Carnosine (1 mM) was found to reduce or prevent the toxic effects of 10-30 µM zinc (Fig. 5, p < 0.001, 10 µM not shown). However, carnosine at 1 mM was not sufficient to prevent the toxicity produced by 100 µM zinc (0.05 < p < 0.1, not shown). In these experiments, investigators used 30 µM copper and varied the concentration of carnosine from 10 µM to 10 mM. Carnosine at concentrations of 1 mM or higher was found to provide significant protection from copper-mediated toxicity (Fig. 5, p < 0.02). However, at concentrations of 10 or 100 µM, carnosine did not significantly protect against copper-mediated toxicity (0.1 < p < 0.2, not shown).
These results [30] contrast with the neurotoxicity data previously reported by Koh and colleagues [72]. Koh et al. have reported that the toxic effects of zinc, but apparently not copper, can be minimized by the co-administration of Ca-EDTA to cultured cortical neurons. However, these two sets of results are difficult to compare, since Koh and colleagues [72] exposed neurons to zinc for 15 min, but used 48 h exposures for copper. No direct comparisons between zinc and copper were reported using the same exposure durations. Nevertheless, the Koh et al. results suggest that chelation alone is insufficient to prevent the toxic actions of copper, since EDTA has a 100-fold higher binding affinity for copper than for zinc. However, the lack of protection afforded by Koh and colleagues' in vivo administration of Zn-EDTA (a saturated form of EDTA that can bind no further zinc) prior to ischemia suggests that chelation does play some role in preventing metal-induced neurotoxicity. Along with their own findings, Koh and colleagues' findings led Horning and colleagues to hypothesize that Ca-EDTA, and potentially carnosine, would afford some degree of protection against both zinc- and copper-mediated toxicity. Therefore, they exposed cultured olfactory bulb neurons to either metal (100-200 µM) with co-applied 1 mM Ca-EDTA for 1 h. In contrast to the Koh et al. result, Ca-EDTA was found to be highly neuroprotective against both zinc- and copper-mediated toxicity for 1 h exposure durations (p < 0.001, data not shown). However, with a 24 h exposure time, 1 mM Ca-EDTA effectively protected against zinc-mediated (p < 0.001) but not copper-mediated (0.4 < p < 0.5) toxicity (data not shown). Furthermore, the co-application of 1 mM carnosine (in contrast to Ca-EDTA) with zinc or copper did not provide significant neuroprotection against either zinc- (p > 1.0) or copper-mediated (p > 0.5) toxicity (data not shown).
MECHANISMS OF ACTION AND FUNCTIONAL IMPLICATIONS
For many years, some investigators considered carnosine to be the leading candidate for the transmitter used by olfactory sensory neurons. However, most investigators report little, if any, effect of carnosine on olfactory bulb neurons, casting doubt on its role as a neurotransmitter (e.g., [20-22]). There is now strong electrophysiological and pharmacological evidence that glutamate is the olfactory sensory neuron transmitter [23]. Furthermore, as already described, even millimolar concentrations of carnosine do not evoke a membrane current in either M/T cells or interneurons in culture [19]. Therefore, most investigators have concluded that carnosine does not function as a neurotransmitter in the olfactory bulb.
Alternatively, some investigators have proposed that carnosine acts as a modulator [1]. This review has already described evidence for the colocalization of zinc/copper and carnosine in olfactory sensory neuron terminals [3, 8] as well as recent evidence that carnosine modulates the effects of transition metals on amino acid receptors and synaptic transmission [19]. However, underlying mechanisms by which colocalized neuroactive substances interact in the olfactory bulb are unclear. Such evidence comes from other systems. For example, it has been demonstrated in the hippocampus that low frequency (1 Hz) stimulation of the perforant path releases glutamate, but fails to release zinc. In contrast, higher frequency (10 Hz) stimulation causes substantial release of glutamate and zinc from the mossy fibers [99]. Thus, activity-dependent modulation of glomeruli could occur if zinc (and/or copper), carnosine, and glutamate were released by different patterns of activity, potentially contributing to odor information processing, synaptic plasticity, or olfactory learning.
The finding that zinc is neurotoxic to cultured olfactory bulb neurons [30] is consistent with the results of previous experiments examining the effects of zinc on hippocampal and cortical neurons. These results also extend prior observations by demonstrating that short-term (1 h) exposure to copper also is neurotoxic [1], and that both zinc and copper can be neurotoxic at physiologically relevant concentrations [30]. Together these findings suggest the likely existence of endogenous neuroprotective mechanisms that prevent normal concentrations of zinc and copper from exerting toxic effects. The finding that carnosine can provide neuroprotection from the toxic actions of both zinc and copper [30] suggests that carnosine, an endogenous dipeptide, may perform such a function. There are a number of means by which transition metals are thought to cause neurotoxicity, thus several possible mechanisms by which carnosine may protect against these effects.
Extracellular zinc may gain access to intracellular compartments through NMDA receptors [100], calcium-permeable AMPA receptors [101], or voltage-gated calcium channels [102]. After entering the cell, zinc can trigger widespread disruptions of normal cellular functions. Zinc has been proposed to disrupt calcium homeostasis [102], inhibit mitochondrial electron transport [52], disrupt tubulin assembly [103], and overactivate calcium-mediated enzymes [104]. Furthermore, zinc reacts with the thiol and imidazole moieties of many proteins, and, thus, can disrupt their structure and function [105].
Although little is known about the biochemical basis underlying copper-mediated neurotoxicity, high concentrations of copper have been shown to interfere with neuronal calcium channels [106], inhibit the enzymatic activity of Na,K-ATPase [107], and bind to proteins, potentially altering their structure and function [105]. Furthermore, copper can enhance neurotoxicity indirectly by generating oxyradicals. Copper ions (Cu(II)) target thiol groups which are capable of reducing Cu(II) to Cu(I). Cu(I) can then be re-oxidized back to the Cu(II) form in the presence of molecular oxygen. Subsequently, molecular oxygen is converted to superoxide radicals that ultimately can lead to lipoperoxidation [108]. In contrast to copper, not only is zinc not redox active and unlikely to generate reactive oxygen species [109], but it may have some antioxidant properties [110].
In almost all studies that identified zinc in a particular location in the brain, or as mediating a particular function (e.g., neurotoxicity), the data were generated by techniques that were much more sensitive to copper. Therefore, some findings attributed to the actions of zinc may actually be due to endogenous copper, or possibly, a combination of zinc and copper. Further studies using more selective techniques will be required to further elucidate the relative roles of zinc and copper in both normal and pathological processes.
The means by which carnosine reduces metal-mediated toxicity are unclear, but several possibilities exist. For example, the chelating ability of the histidine residue of carnosine [27] may contribute to its attenuation of zinc- and copper-mediated toxicity. The demonstrated ability of carnosine to function as an antioxidant (e.g., [26, 28]) also may have contributed to the reported decrease in neuronal death [30]. Furthermore, both zinc [74] and copper [111] have been implicated in the aggregation of betaA, the primary component of Alzheimer-associated beta-amyloid deposits. The concentrations of zinc and copper are significantly elevated within the neuropil of patients with Alzheimer's disease, and even higher concentrations are found within the core and periphery of plaque deposits [112]. Recent evidence suggests that carnosine can inhibit the aggregation of amyloid peptide [113], and that toxic effects of amyloid peptide can be prevented or reduced by carnosine [114]. Experiments are currently underway to further explore these relationships.
In conclusion, carnosine appears to have little or no direct effect on olfactory bulb neurons, amino acid receptors, or synaptic transmission. However, recent results suggest that carnosine may act indirectly as a neuromodulator by influencing the effects of zinc and copper on neuronal excitability.
Furthermore, physiologically relevant concentrations of zinc and copper also can be neurotoxic, and carnosine can reduce or prevent zinc- or copper-mediated neurotoxicity. Understanding the role of endogenous compounds with putative neuromodulatory and/or neuroprotective actions, such as carnosine, may help in the development of clinical approaches for the treatment of neuropathologies that involve metals and free radicals.
REFERENCES
1.Biffo, S., Grillo, M., and Margolis, F. L. (1990)
Neuroscience, 35, 637-651.
2.Pognetto, M. S., Cantino, D., and Fasolo, A. (1992)
Brain Res., 578, 261-268.
3.Sassoe-Pognetto, M., Cantino, D., Panzanelli, P.,
di Cantogno, L. V., Giustetto, M., Margolis, F. L., de Biasi, S., and
Fasolo, A. (1993) Neuroreport, 5, 7-10.
4.Bauer, K., Hallermayer, K., Salnikow, J.,
Kleinkauf, H., and Hamprecht, B. (1982) J. Biol. Chem.,
257, 3593-3597.
5.Bakardjiev, A. (1997) Neurosci. Lett.,
227, 115-118.
6.Sakai, M., Kani, K., Karasawa, N., Yoshida, M., and
Nagatsu, I. (1988) Brain Res., 438, 335-338.
7.Ferriero, D., and Margolis, F. L. (1975) Brain
Res., 94, 75-86.
8.Friedman, B., and Price, J. L. (1984) J. Comp.
Neurol., 223, 88-109.
9.Ashihara, M., Ohmori, T., Nishimura, T., Sakai, M.,
and Nagatsu, I. (1993) Acta Otolaryngol. Suppl. (Stockholm),
506, 34-36.
10.Harding, J. W., and O'Fallon, J. V. (1979)
Brain Res., 173, 99-109.
11.Burd, G. D., Davis, B. J., Macrides, F., Grillo,
M., and Margolis, F. L. (1982) J. Neurosci., 2,
244-255.
12.Schulz, M., Hamprecht, B., Kleinkauf, H., and
Bauer, K. (1987) J. Neurochem., 49, 748-755.
13.Yamashita, T., Shimada, S., Guo, W., Sato, K.,
Kohmura, E., Hayakawa, T., Takagi, T., and Tohyama, M. (1997) J.
Biol. Chem., 272, 10205-10211.
14.Rochel, S., and Margolis, F. L. (1982) J.
Neurochem., 38, 1505-1514.
15.Bakardjiev, A. (1998) Glia, 24,
346-351.
16.Hirsch, J. D., Grillo, M., and Margolis, F. L.
(1978) Brain Res., 158, 407-422.
17.Hirsch, J. D., and Margolis, F. L. (1979)
Brain Res. (Biochem. Biol. Stud.), 174, 81-94.
18.Kanaki, K., Kawashima, S., Kashiwayanagi, M., and
Kurihara, K. (1997) Neurosci. Lett., 231, 167-170.
19.Trombley, P. Q., Horning, M. S., and Blakemore,
L. J. (1998) Neuroreport, 9, 3503-3507.
20.Frosch, M. P., and Dichter, M. A. (1984) Brain
Res., 290, 321-332.
21.Nicoll, R. A., Alger, B. E., and Jahr, C. E.
(1980) Proc. Roy. Soc. Lond. Br. Biol. Sci., 210,
133-149.
22.MacLeod, N. K., and Straughan, D. W. (1979)
Exp. Brain Res., 34, 183-188.
23.Berkowicz, D. A., Trombley, P. Q., and Shepherd,
G. M. (1994) J. Neurophysiol., 71, 2557-2561.
24.Ennis, M., Zimmer, L. A., and Shipley, M. T.
(1996) Neuroreport, 7, 989-992.
25.Aroniadou-Anderjaska, V., Ennis, M., and Shipley,
M. T. (1997) Neuroscience, 79, 425-434.
26.Boldyrev, A. A., Stvolinsky, S. L., Tyulina, O.
V., Koshelev, V. B., Hori, N., and Carpenter, D. O. (1997) Cell.
Mol. Neurobiol., 17, 259-271.
27.Brown, C. E., and Antholine, W. E. (1979) J.
Phys. Chem., 83, 3314-3319.
28.Kohen, R., Yamamoto, Y., Cundy, K. C., and Ames,
B. N. (1988) Proc. Natl. Acad. Sci. USA, 85,
3175-3179.
29.Stvolinsky, S. L., Kukley, M. L., Dobrota, D.,
Matejovicova, M., Tkac, I., and Boldyrev, A. A. (1999) Cell. Mol.
Neurobiol., 19, 45-56.
30.Horning, M. S., Blakemore, L. J., and Trombley,
P. Q. (2000) Brain Res., in press.
31.Cuajungco, M. P., and Lees, G. J. (1997) Brain
Res. (Brain Res. Rev.), 23, 219-236.
32.Frederickson, C. J. (1989) Int. Rev.
Neurobiol., 31, 145-238.
33.Ebadi, M. (1991) Meth. Enzymol.,
205, 363-387.
34.Itoh, M., Ebadi, M., and Swanson, S. (1983) J.
Neurochem., 41, 823-829.
35.Ebadi, M., and Hama, Y. (1986) Adv. Exp. Med.
Biol., 203, 557-570.
36.Simons, T. J. (1991) J. Membr. Biol.,
123, 63-71.
37.Reyes, J. G. (1996) Am. J. Physiol.,
270, C401-410.
38.Danscher, G. (1981) Histochemistry,
71, 1-16.
39.Danscher, G. (1996) Histochem. J.,
28, 361-373.
40.Danscher, G., Howell, G., Perez-Clausell, J., and
Hertel, N. (1985) Histochemistry, 83, 419-422.
41.Perez-Clausell, J., and Danscher, G. (1985)
Brain Res., 337, 91-98.
42.Frederickson, C. J., Klitenick, M. A., Manton, W.
I., and Kirkpatrick, J. B. (1983) Brain Res., 273,
335-339.
43.Slomianka, L., Danscher, G., and Frederickson, C.
J. (1990) Neuroscience, 38, 843-854.
44.Masters, B. A., Quaife, C. J., Erickson, J. C.,
Kelly, E. J., Froelick, G. J., Zambrowicz, B. P., Brinster, R. L., and
Palmiter, R. D. (1994) J. Neurosci., 14, 5844-5857.
45.Frederickson, C. J., Hernandez, M. D., and
McGinty, J. F. (1989) Brain Res., 480, 317-321.
46.Beaulieu, C., Dyck, R., and Cynader, M. (1992)
Neuroreport, 3, 861-864.
47.Assaf, S. Y., and Chung, S. H. (1984)
Nature, 308, 734-736.
48.Hartter, D. E., and Barnea, A. (1988)
Synapse, 2, 412-415.
49.Kardos, J., Kovacs, I., Hajos, F., Kalman, M.,
and Simonyi, M. (1989) Neurosci. Lett., 103, 139-144.
50.Trombley, P. Q., and Shepherd, G. M. (1996) J.
Neurophysiol., 76, 2536-2546.
51.Gulya, K., Kovacs, G. L., and Kasa, P. (1991)
Life Sci., 48, PL57-62.
52.Donaldson, J., Pierre, T. S., Minnich, J. L., and
Barbeau, A. (1973) Can. J. Biochem., 51, 87-92.
53.Bradford, H. F. (1986) Chemical
Neurobiology, Freeman, N. Y.
54.Dreosti, I. E. (1983) in Neurobiology of the
Trace Elements: Trace Element Neurobiology and Deficiencies
(Dreosti, I. E., and Smith, R. M., eds.) Vol. 1, Humana Press, New
Jersey, pp. 135-162.
55.Howell, G. A., Welch, M. G., and Frederickson, C.
J. (1984) Nature, 308, 736-738.
56.Xie, X. M., and Smart, T. G. (1991)
Nature, 349, 521-524.
57.Xie, X., and Smart, T. G. (1993) Eur. J.
Neurosci., 5, 430-436.
58.Xie, X., Hider, R. C., and Smart, T. G. (1994)
J. Physiol., 478, 75-86.
59.Forsythe, I. D., Westbrook, G. L., and Mayer, M.
L. (1988) J. Neurosci., 8, 3733-3741.
60.Mayer, M. L., and Vyklicky, L., Jr. (1989) J.
Physiol. (Lond), 415, 351-365.
61.Westbrook, G. L., and Mayer, M. L. (1987)
Nature, 328, 640-643.
62.Smart, T. G. (1992) J. Physiol.,
447, 587-625.
63.Sato, M., Ohtomo, K., Daimon, T., Sugiyama, T.,
and Iijima, K. (1994) J. Histochem. Cytochem., 42,
1585-1591.
64.Vlachova, V., Zemkova, H., and Vyklicky, L., Jr.
(1996) Eur. J. Neurosci., 8, 2257-2264.
65.Weiser, T., and Wienrich, M. (1996) Brain
Res., 742, 211-218.
66.Xiong, K., Peoples, R. W., Montgomery, J. P.,
Chiang, Y., Stewart, R. R., Weight, F. F., and Li, C. (1999) J.
Neurophysiol., 81, 2088-2094.
67.Kosaka, T., Kosaka, K., Heizmann, C. W., Nagatsu,
I., Wu, J., Yanaihara, N., and Hama, K. (1987) Brain Res.,
411, 373-378.
68.Trombley, P. Q., and Shepherd, G. M. (1994) J.
Neurophysiol., 71, 761-767.
69.Van den Pol, A. N., and Gorcs, T. (1988) J.
Neurosci., 8, 472-492.
70.Frederickson, C. J., Hernandez, M. D., Goik, S.
A., Morton, J. D., and McGinty, J. F. (1988) Brain Res.,
446, 383-386.
71.Tonder, N., Johansen, F. F., Frederickson, C. J.,
Zimmer, J., and Diemer, N. H. (1990) Neurosci. Lett.,
109, 247-252.
72.Koh, J. Y., Suh, S. W., Gwag, B. J., He, Y. Y.,
Hsu, C. Y., and Choi, D. W. (1996) Science, 272,
1013-1016.
73.Bush, A. I., Moir, R. D., Rosenkranz, K. M., and
Tanzi, R. E. (1995) Science, 268, 1921-1923.
74.Bush, A. I., Pettingell, W. H., Multhaup, G.,
Paradis, M., Vonsattel, J. P., Gusella, J. F., Beyreuther, K., Masters,
C. L., and Tanzi, R. E. (1994) Science, 265,
1464-1467.
75.Clements, A., Allsop, D., Walsh, D. M., and
Williams, C. H. (1996) J. Neurochem., 66, 740-747.
76.Esler, W. P., Stimson, E. R., Jennings, J. M.,
Ghilardi, J. R., Mantyh, P. W., and Maggio, J. E. (1996) J.
Neurochem., 66, 723-732.
77.Mantyh, P. W., Ghilardi, J. R., Rogers, S.,
DeMaster, E., Allen, C. J., Stimson, E. R., and Maggio, J. E. (1993)
J. Neurochem., 61, 1171-1174.
78.Gschwind, M., and Huber, G. (1995) J.
Neurochem., 65, 292-300.
79.Koh, J. Y., Yang, L. L., and Cotman, C. W. (1990)
Brain Res., 533, 315-320.
80.Mattson, M. P., Cheng, B., Davis, D., Bryant, K.,
Lieberburg, I., and Rydel, R. E. (1992) J. Neurosci., 12,
376-389.
81.Blair-West, J. R., Denton, D. A., Gibson, A. P.,
and McKinley, M. J. (1990) Brain Res., 507, 6-10.
82.Fliss, H., and Menard, M. (1991) Arch.
Biochem. Biophys., 287, 175-179.
83.Fliss, H., and Menard, M. (1992) Arch.
Biochem. Biophys., 293, 195-199.
84.Fliss, H., Menard, M., and Desai, M. (1991)
Can. J. Physiol. Pharmacol., 69, 1686-1691.
85.Maret, W. (1995) Neurochem. Int.,
27, 111-117.
86.Messori, L., Poggetto, G. D., Monnanni, R., and
Hirose, J. (1997) Biometals, 10, 303-313.
87.Rosen, D. R., Siddique, T., Patterson, D.,
Figlewicz, D. A., Sapp, P., Hentati, A., Donaldson, D., Goto, J.,
O'Regan, J. P., Deng, H. X., et al. (1993) Nature, 362,
59-62.
88.Constantinidis, J., and Tissot, R. (1981) Adv.
Biochem. Psychopharmacol., 27, 413-422.
89.DiDonato, M., and Sarkar, B. (1997) Biochim.
Biophys. Acta, 1360, 3-16.
90.Multhaup, G. (1997) Biomed. Pharmacother.,
51, 105-111.
91.Tumer, Z., and Horn, N. (1997) J. Med.
Genet., 34, 265-274.
92.Ferreyra, M. H., and Barragan, E. (1989) Int.
J. Neurosci., 49, 157-197.
93.Doty, R. L. (1991) Ann. N. Y. Acad. Sci.,
640, 20-27.
94.Talamo, B. R., Rudel, R., Kosik, K. S., Lee, V.
M., Neff, S., Adelman, L., and Kauer, J. S. (1989) Nature,
337, 736-739.
95.Samudralwar, D. L., Diprete, C. C., Ni, B. F.,
Ehmann, W. D., and Markesbery, W. R. (1995) J. Neurol. Sci.,
130, 139-145.
96.Thompson, C. M., Markesbery, W. R., Ehmann, W.
D., Mao, Y. X., and Vance, D. E. (1988) Neurotoxicology,
9, 1-7.
97.Corrigan, F. M., Reynolds, G. P., and Ward, N. I.
(1993) Biometals, 6, 149-154.
98.Wenstrup, D., Ehmann, W. D., and Markesbery, W.
R. (1990) Brain Res., 533, 125-131.
99.Aniksztejn, L., Charlton, G., and Ben-Ari, Y.
(1987) Brain Res., 404, 58-64.
100.Choi, D. W., Yokoyama, M., and Koh, J. (1988)
Neuroscience, 24, 67-79.
101.Yin, H. Z., and Weiss, J. H. (1995)
Neuroreport, 6, 2553-2556.
102.Weiss, J. H., Hartley, D. M., Koh, J. Y., and
Choi, D. W. (1993) Neuron, 10, 43-49.
103.Kress, Y., Gaskin, F., Brosnan, C. F., and
Levine, S. (1981) Brain Res., 220, 139-149.
104.Csermely, P., Szamel, M., Resch, K., and
Somogyi, J. (1988) J. Biol. Chem., 263, 6487-6490.
105.Chvapil, M., Ryan, J. N., and Zukoski, C. F.
(1972) Proc. Soc. Exp. Biol. Med., 140, 642-646.
106.Schulte, S., Muller, W. E., and Friedberg, K.
D. (1995) Toxicology, 97, 113-121.
107.Li, J., Lock, R. A., Klaren, P. H., Swarts, H.
G., Schuurmans Stekhoven, F. M., Wendelaar Bonga, S. E., and Flik, G.
(1996) Toxicol. Lett., 87, 31-38.
108.Agarwal, K., Sharma, A., and Talukder, G.
(1989) Chem. Biol. Interact., 69, 1-16.
109.Berg, J. M., and Shi, Y. (1996) Science,
271, 1081-1085.
110.Bray, T. M., and Bettger, W. J. (1990) Free
Radic. Biol. Med., 8, 281-291.
111.Atwood, C. S., Moir, R. D., Huang, X., Scarpa,
R. C., Bacarra, N. M., Romano, D. M., Hartshorn, M. A., Tanzi, R. E.,
and Bush, A. I. (1998) J. Biol. Chem., 273,
12817-12826.
112.Lovell, M. A., Robertson, J. D., Teesdale, W.
J., Campbell, J. L., and Markesbery, W. R. (1998) J. Neurol.
Sci., 158, 47-52.
113.Munch, G., Mayer, S., Michaelis, J., Hipkiss,
A. R., Riederer, P., Muller, R., Neumann, A., Schinzel, R., and
Cunningham, A. M. (1997) Biochim. Biophys. Acta, 1360,
17-29.
114.Preston, J. E., Hipkiss, A. R., Himsworth, D.
T., Romero, I. A., and Abbott, J. N. (1998) Neurosci. Lett.,
242, 105-108.