Vascular Smooth Muscle Actions of Carnosine as Its Zinc Complex Are Mediated by Histamine H1 and H2 Receptors
D. J. Miller* and A. O'Dowd
Clinical Research Initiative in “Heart Failure”, Institute of Biomedical and Life Sciences, West Medical Building, University of Glasgow, Glasgow G12 8QQ, Scotland, UK; E-mail: D.J.Miller@bio.gla.ac.uk* To whom correspondence should be addressed.
Received March 20, 2000
The endogenous dipeptide carnosine (beta-alanyl-L-histidine), at 0.1-10 mM, can provoke sustained contractures in rabbit saphenous vein rings with greater efficacy than noradrenaline. The effects are specific; anserine and homocarnosine are ineffective, as are carnosine's constituent amino acids histidine and beta-alanine. Zinc ions enhance the maximum carnosine-induced tension (to127 ± 13% of control at 10 µM Zntotal) and muscle sensitivity is potentiated (mean K0.5 reduced from 1.23 mM to 17 µM carnosine with 15 µM Zntotal). The dipeptide acts as a Zn-carnosine complex (Zn·Carn). The effects of carnosine at 1 µM-10 mM (total) in the presence of 1-100 µM Zn2+ (total) can be described as a unique function of [Zn·Carn] with an apparent K0.5 for the complex of 7.4·10-8 M. Contractures are reduced at low [Ca2+], unaffected by adrenoceptor antagonists, but can be blocked by antagonists to several receptor types. The most specific effect is by mepyramine, the H1 receptor antagonist. With Zn present, carnosine can inhibit the H1-specific binding of [3H]mepyramine to isolated Guinea pig cerebellar membranes. This effect of carnosine can be described as a function of the concentration of Zn·Carn with an apparent IC50 of 2.45 µM. Like histamine, carnosine evoked an H2-mediated (cimetidine-sensitive) relaxation in the presence of mepyramine, but was less potent (10.8 ± 3.1% of initial tension remaining at 10 mM carnosine compared with 13.4 ± 7.5% remaining at 0.1 mM histamine). Preliminary studies with a Zn-selective fluorescent probe indicate that functionally significant levels of Zn can be released from adventitial mast cells that could modulate actions of carnosine in the extravascular space as well as those of histamine itself. We conclude that carnosine can act at the smooth muscle H1-receptor to provoke vasoconstriction and that it also has the potential to act at H1-receptors in the central nervous system. Carnosine's mode of action is virtually unique: a vascular muscle receptor apparently transduces the action of a dipeptide in the form of a metal chelate. The functional relationship of carnosine with histamine and the possible physiological relevance of Zn ions for the activity of both agents have not previously been reported.
KEY WORDS: carnosine, zinc, vascular smooth muscle, histamine receptors
Our studies into the physiological actions of carnosine stemmed from seeking an “imidazole-containing” structure amongst the components of the sarcoplasm. At an early stage, we were interested to find whether any endogenous “imidazoles” could have properties similar to the myofilament Ca2+-sensitizing then recently first detected for caffeine. Even earlier work by one of us [1] led us to consider that the “imidazole” moiety in the methylxanthines such as caffeine (and its relatives such as theophylline and theobromine) was the likely key to this activity.
The histidyl dipeptide carnosine (beta-alanyl-L-histidine), one of a group of endogenous compounds including anserine (beta-alanyl-N1-methyl-L-histidine) and homocarnosine (gamma-aminobutyryl-L-histidine) seemed a candidate [2]. Carnosine occurs at high concentration in skeletal muscle (1-50 mM) [3] and figures as the most prevalent of the organic constituents of myoplasm at least in fast skeletal muscle. Its concentration is lower in cardiac muscle (~1 mM) [4] and in plasma the concentration is in many cases undetectable (<0.1 µM by the most sensitive method to date) [5]. Amongst the actions attributed to it (see [6]) carnosine has been implicated as: a) major organic pH buffer of skeletal muscle; b) having radical scavenging activity (see, e.g., [7]); c) an olfactory neurotransmitter and/or a neuromodulator [8]; d) influencing at least two enzymes important in the energy transduction pathway, phosphorylase a and b [6]. We added its Ca2+-sensitizing action in skeletal and cardiac muscle to this list [2, 4, 9]. To introduce the most recent target of our attention, it has been known for nearly seventy years to have effects on vascular tone [10, 11]. Those papers reported that bolus injections of carnosine in vivo reduce blood pressure, although this action remains unexplained. Consistent with these reports, we have found that carnosine constricts isolated blood vessels. Unusually for any “ligand”, the dipeptide acts in the form of a Zn-carnosine complex (Zn·Carn) at micromolar concentration. This is a receptor-mediated action.
In this paper, we review the key findings that led us to our present understanding of carnosine's vascular actions. We report some new findings that give clues to the possible physiological origin of the Zn ions apparently crucial to carnosine action in vascular smooth muscle. We believe these findings help to understand how this fascinating dipeptide works and why its actions have remained experimentally unpredictable or elusive. They also suggest that other, more widely known agonists might be linked in previously unsuspected ways with that of carnosine.
MATERIALS AND METHODS
The study of vascular preparations was by the conventional “vascular ring” technique. The methods have been described in detail elsewhere [12, 13]. In outline, vascular rings (2-3 mm long) from the lateral saphenous vein from the rabbit (New Zealand White, 2.2-2.4 kg, sacrificed by intravenousoverdose of sodium pentobarbitone) were mounted for isometric tension recording in standard organ baths (10-15 ml) containing normal physiological saline (118.4 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 1.2 mM MgSO4, 24.9 mM NaHCO3, 1.2 mM KH2PO4, 11.1 mM D-glucose) at 37°C equilibrated with 95% oxygen and 5% CO2.
Protocol. Preparations were mounted under low resting tension and allowed to relax before setting an initial resting tension of 0.3-0.5 g wt. During a 1 h equilibration time, bathing solutions were changed once, followed by a further 1 h for equilibration. Zn ions were applied as zinc acetate, added 5 to 10 min before the addition of carnosine. The same initial protocol was employed in experiments involving the various antagonists except that tissues were pre-exposed to a single dose of carnosine (10-4 M) during the second equilibration period in order to minimize changes in sensitivity between first and second cumulative exposures to carnosine. Preparations were then generally cumulatively activated (10-7 to 3·10-3 M carnosine). After washout, preparations were incubated for 30 min with the antagonist and the cumulative exposure to carnosine repeated. Several preparations from the same animal were usually assayed in parallel with one as a time control. Antagonist experiments were all done in the presence of 5 µM Zn2+ (to ensure a stable, substantial response to carnosine). In the histamine experiments, all tissues were pre-exposed to a single submaximal dose of histamine (3·10-5 M) or carnosine (10-4 M in the presence of 5 µM Zn2+) before the main protocol was started.
Contractures were evoked by applying the agonist from stock (at
pH 7.4 where relevant) at 0.1% of bath volume. Drugs were added in
the same way. Tension was allowed to stabilize (see Fig. 1a) before agonist concentration was raised further.
Tension signals were recorded (usually at 5 Hz) and analyzed using
MacLab 8 (A-D Instruments) hardware and software. Steady state
tension at each agonist concentration was measured. Dose-response
curves were fitted to the Hill equation:
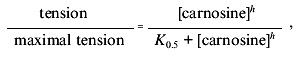
Some preliminary observations were made on small, subcutaneous “resistance” arterioles (~200-300 µm in diameter) by wire myography (see [14] for details). The protocols and solutions used were identical to those for the venous rings.
Relaxation experiments. Preparations were pre-contracted with noradrenaline (NA) in the presence or absence of mepyramine (10-5 M), which was added 20 min before NA exposure. When the tension had stabilized, histamine or carnosine was applied cumulatively, followed by cimetidine (10-5 M). In a different protocol, carnosine (10-3 to 10-2 M) was used as the contractile agent (no mepyramine present) before the addition of histamine.
Fluorescence microscopy of Zn2+ release. In four preliminary experiments, short lengths of rabbit saphenous were viewed by confocal microscopy. We used the new, Zn-fluorescent, cell-impermeant probe Newport Green 7990 (Molecular Probes, Inc., USA). This is a single wavelength dye that increases fluorescence (peak at 530 nm) upon binding Zn ions, with a Kd of about 1 µM under physiological ionic conditions. With excitation at 488 nm, we confirmed that a [Zn2+] fluorescence signal is readily detectable above 0.1 µM. The modest sensitivity of the dye to Ca2+ we found to be adequately avoided by lowering saline [Ca2+] to 0.2 mM; the dye proves insensitive to Mg2+. We have imaged the plane of putative mast cells in the adventitia where the nuclei were stained with a vital dye Hoechst 33342 (Hoechst, Germany) rendering them visible (under 364 nm UV illumination), which makes recognition of the adventitia, medial, and endothelial layers straightforward. The field of view employed was approximately 80 × 80 µm and included seven or eight mast cell nuclei. After applying the Newport dye, a signal was detected which stabilized within 5 min (mostly attributable to the background 0.2 mM Ca2+ in the saline). The light level was sustained for many minutes unless interventions were made.
The depth of field was increased (“confocality” thereby reduced) to several micrometers to minimize effects of small movement artifacts on the putative Zn signal. Light was collected for 10 sec every 15 sec and digitized images were recorded for off-line analysis. Regions were chosen in apparently featureless extracellular space from the recorded image sequence to provide several intensity-versus-time traces, one of which is plotted in Fig. 5. Mast cell degranulation was provoked by injecting Compound 48/80 into the microscope slide bath to give a final concentration of 1 or 10 µM for each bolus. In some cases, EDTA was applied in the same way (to 10 µM) or was present (at 50 µM) in the bathing solution throughout.
Calculations of Zn-carnosine complex concentration. The concentration of the Zn-carnosine complex (Zn·Carn) reported in Fig. 3 was calculated using a multi-ligand, multi-ion program (REACT, by Prof. G. L. Smith, University of Glasgow) according to principles described elsewhere [15] developed for work with chemically “skinned” muscle fibers. The relevant stoichiometric affinity constants were employed [16, 17]. Values for the associations for carnosine (L), LH·Ca and LH·Mg, are not available in the literature, but may be estimated pro rata to approximate 1.0 (log10K). However, trial calculations revealed that [Zn·Carn] was hardly affected by these reactions. The same tabulation was used for the relevant EDTA constants. Solution pHactivity was assumed to be 7.4 (bicarbonate/CO2 buffered at 37°C).
Mean data ± s.e.m. for accumulated and derived parameters are quoted as n (number of preparations and/or number of animals). The magnitude of the effects to be demonstrated are mostly so great that simple inspection reveals the significance (means differ by more than several times the sem). In some cases, Student's t-test has been applied (comparing populations as “test” versus “control” at a given carnosine concentration) to test significance.
RESULTS
Our preliminary work established that a small proportion of vessel preparations respond to 10-100 µM carnosine with a sustained contracture but widely varying amplitude (relative to, e.g., a standard challenge with maximal effective concentration of noradrenaline (NA)). Additionally, we found a small, but significant, tendency of carnosine to potentiate contractures induced by NA. Skeletal muscle intracellular [carnosine] can exceed 10 mM, so we considered that high concentrations might occur at certain times in the extracellular space. Carnosine consistently evoked rapid, sustained at 0.1 mM or more, effect (Fig. 1a). One striking feature is that the maximum tension evoked by carnosine is approximately double that for an optimal concentration of NA (e.g., Fig. 1b). A concentration-response curve for carnosine (obtained cumulatively) can thereby be obtained (Fig. 1c). Repeat challenges show slight desensitization occurring, typical of many agonists in isolated blood vessels (see [12]). Our protocols thus routinely compare between the second sets of responses. Otherwise, the responses to carnosine are reproducible and reversible although “control” sensitivity to carnosine between groups of experiments can vary significantly. These contractures do not require functional endothelium (removed by “rubbing” the vessel) and, with the endothelium intact, acetylcholine promotes rapid, full relaxation of the carnosine-induced contractures (CICs). Unlike carnosine, anserine, homocarnosine (up to 1 mM) and carnosine's constituent amino acids histidine (to 1 mM) and beta-alanine (to 10 mM) are all ineffective.
Studies made with NA routinely employ a low concentration of EDTA in order to chelate contaminant iron ions and thereby minimize NA oxidation. We found EDTA (1-100 µM) attenuates the CIC (e.g., Fig. 1a) in a dose-dependent way. This immediately suggested that a trace heavy metal ion was somehow essential for the action of carnosine (note that 20 µM EDTA has only negligible effects on [Ca2+] or [Mg2+], which are present in considerable “excess” in the salines). Carnosine binds a number of ions including Cu2+, Mn2+, Fe2+, Ni2+, and Zn2+ [16], some of which might be plausible, “physiologically-relevant” candidates, so we tested each of them. Up to 10 µM [metal ion]total, only Zn facilitated carnosine's action. Several of the other heavy metals are well known as blockers of Ca2+-channels and thus tend to inhibit contraction in blood vessels. Furthermore, Zn2+ alone, or the accompanying anion, acetate, do not induce contracture without carnosine being present. This strongly suggested that Zn2+ ions are specifically involved in carnosine action. To provide an initial characterization, we studied the effects of 15 µM Zn2+, the total concentration reported in mammalian plasma [18], in more detail. The effect was dramatic since including Zn2+ reduced the mean K0.5 from 1.23 mM (29 preparations, 11 animals) to 17.0 µM (12 preparations, 7 animals). The apparent threshold for carnosine-induced contractures was now reduced to near 1 µM and maximum tension was achieved at 100 µM. The rightward shift produced by EDTA is partially reversed by adding 15 µM Zn2+.Fig. 1. Basic actions of carnosine on contraction of saphenous vein: a) cumulative response to carnosine (1) is largely blocked by 20 µM EDTA (2); b) relative effect of optimal noradrenaline concentration (30 µM) (1) is exceeded by that of 10 mM carnosine (2); c) normalized concentration-response curve (mean ± s.e.m. for 12 preparations and 7 animals) obtained with carnosine (5 µM Zn present). Maximal contracture in the presence of carnosine alone is 3.03 ± 0.11 g.wt. (1) and that in the presence of noradrenaline alone is 1.60 ± 0.31 g.wt. (2).
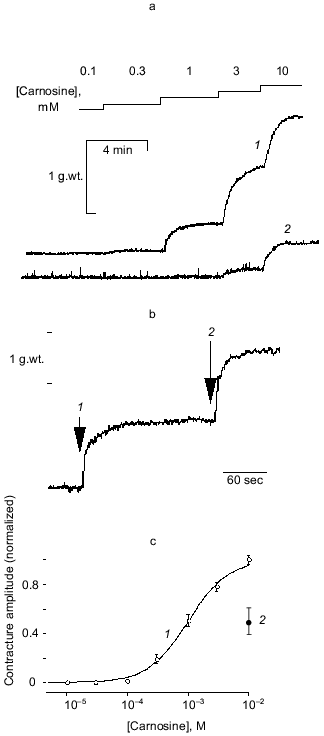
Characterization of zinc-carnosine interaction.Chelation of any Zn2+ present in the nominally Zn2+-free conditions by EDTA would prevent Zn2+-carnosine complex formation. We hypothesized, therefore, that carnosine might act in the form of a Zn2+-carnosine complex. This hypothesis was tested by using a range of Zn2+ concentrations (1 to 100 µM added). The results are most illuminatingly considered when plotted as contracture response versus the calculated concentration of the Zn·Carn complex (Fig. 2)--“carnosine-induced” contractures can best be understood as “Zn·Carn-induced” contractures. Regardless of absolute concentrations of either Zn2+ or carnosine (each over a 100-fold range), the data can be collapsed onto a single curve. This previously unreported agonist (Zn·Carn) is effective at plausibly low concentrations in the range of 10-8 to 10-6 M with EC50 = 7.4·10-8 M [12].
Even the results obtained in nominally Zn-free conditions, or in the presence of EDTA, can be satisfactorily explained by assuming a specific “contamination” level of Zn (either in the media or derived from the preparation) of as little as 0.25 µM Zn contamination. Atomic absorption spectrophotometry of our solutions reports 0.5 ± 0.15 µM Zn.Fig. 2. The steady-state tension (mean ± s.e.m. for 4-29 preparations from 2-15 animals) developed in response to 11 different concentrations of carnosine (10-7 to 10-2 M) is plotted against the concentration of Zn·Carn calculated as described in “Materials and Methods” [12]. The curve is fitted with K0.5 of 7.4·10-8 M and a Hill coefficient of 1.34 (solubility limits imply that nominally 100 µM Zntotal was actually approximately 80 µM). Zinc concentrations (µM): 1) 1; 2) 5; 3) 10; 4) 15; 5) 30; 6) 100; 7) control (0.25 µM).
Since most Zn ions are bound to the plasma protein albumin in vivo, we tested the ability of albumin to antagonize carnosine. As far as practical limits to albumin concentration allow, we could show a simple antagonism [12]. In vivo competition with plasma albumin for Zn ions probably keeps circulating [Zn·Carn] near or below the threshold for vasoactivity; however, where albumin is largely excluded (notably interstitial and cerebrospinal fluids) Zn·Carn formation could perhaps assume more importance.
Receptor transduction of carnosine's action? CIC amplitude and form was affected by reducing extracellular [Ca2+] from 2.5 to 0.5 mM or lower (6 preparations, 2 animals); amplitude was reduced (tension was 61% or less at 0.1 mM Ca2+ or less; 7 preparations, 3 animals) and contractures were less well sustained (<0.1 mM) and abolished in nominally zero Ca2+ (3 preparations, 2 animals) but returned when [Ca2+] was restored to normal.
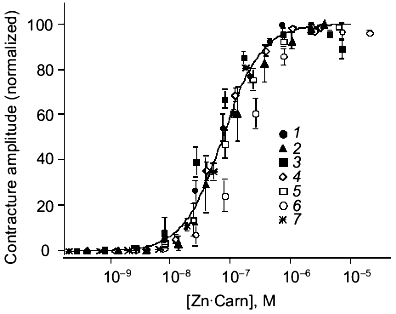
The speed and selectivity of action of carnosine (or apparently Zn·Carn) suggested the involvement of a receptor-mediated pathway. The lack of sensitivity to a range of adrenoreceptor antagonists dismisses that possibility. This was unlikely anyway because the chemistry of catecholamines and the histidyl dipeptide carnosine are very dissimilar. In our first published analysis, we were misled by the apparent sensitivity of the Zn·Carn contracture to a range of 5-HT (5-hydroxytryptamine) antagonists, including ketanserin, cinanserin (see Fig. 3a) and methiothepin [12]. This sensitivity suggested that a “5-HT1-like” receptor [19, 20] might be involved. We had tested histidine as a logical metabolite of carnosine, but found it to be ineffective despite its ability to bind Zn. This initially prevented any consideration of histamine receptors as a possibility, though histamine could be considered the next logical compound related to histidine. However, we subsequently found that the high affinity histamine H1 antagonist mepyramine [21] antagonizes carnosine-induced contractures (see Fig. 3b) at concentrations deemed selective for the H1 receptor (pA2 = 7.97 ± 0.12) [22]. The well established pharmacology of mepyramine suggested very strongly that the H1 receptor must be the one concerned.
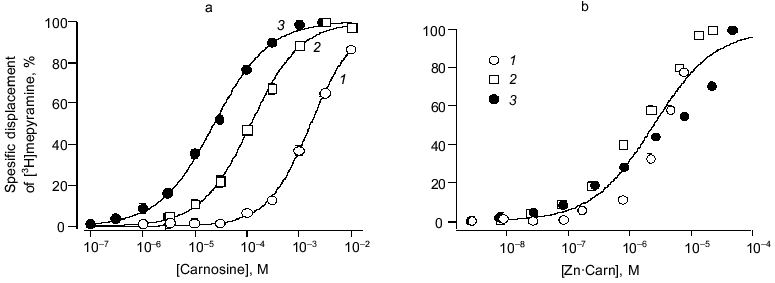
To provide conclusive evidence for our tentative conclusion, we extended these functional observations on blood vessels to study the displacement of tritiated mepyramine from a brain-derived H1 receptor-rich membrane fraction (see Fig. 4). The figure shows the increased inhibition of mepyramine binding by carnosine at three levels of Zn. In each case, the nonspecific displacement attributable to free Zn itself (previously noted by others [23]) was subtracted to leave the carnosine-specific component in this analysis. As previously for the contractile effect on vascular smooth muscle, we “collapsed” these three curves onto a single curve by plotting the data against the calculated concentration of the Zn·Carn complex. The apparent affinity of the brain H1 receptor for Zn·Carn is thus 10-5.61 or 2.45 µM. Whilst this value is lower than that from the functional studies, we do not know the details of the relationship between receptor occupancy and biological effect (in this case, smooth muscle contraction).Fig. 3. Effects of antagonists on carnosine-induced contractures: a) the ability of 1 µM cinanserin (a serotonergic receptor blocker) to reverse the contracture; b) concentration-response curves for the inhibition produced by mepyramine at concentrations of 10 (1) and 100 nM (2); 3) control. Carnosine contractures were induced with 5 µM Zn2+ present throughout.
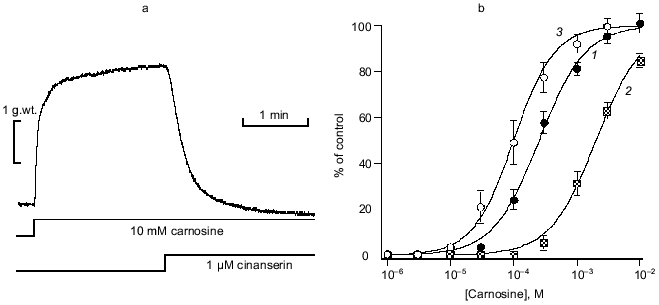
H2 receptor actions of carnosine. To complement this very clear-cut evidence to support the view that carnosine acts in the form of its Zn complex via the histamine H1 receptor to evoke contraction, we made preliminary tests of carnosine with respect to the vascular H2 receptor. We used a protocol of pre-activating the vascular preparations with a near maximal effective dose of noradrenaline (10 µM). In the presence of mepyramine (to block the contractile, H1 mediated effect just described) carnosine now produces substantial relaxation. This relaxing effect, shared with histamine itself, is mediated by an H2 receptor action. This is confirmed by the ability of the selective blocker cimetidine to prevent these actions of both carnosine and histamine. In marked contrast to the H1-mediated actions, however, Zn (5 to 30 µM) had no modifying effect. Thus, we conclude that the role of Zn is specific to the H1 site but, nevertheless, H2 actions by carnosine (albeit at millimolar concentration) can be demonstrated. This may be highly relevant for H2 receptor function in the gastric wall, for example, particularly when high levels of carnosine are ingested in a meat-rich diet. Furthermore, actions at H3 (neuronal) sites have yet to be tested.Fig. 4. Carnosine specific displacement of radiolabeled mepyramine from H1 receptor-enriched guinea pig cerebellar membranes. a) The effect of carnosine at three concentrations of Zntotal (µM): 1) 10; 2) 30; 3) 80. b) The same data now plotted against the calculated concentration of the Zn·Carn complex (see “Materials and Methods” and text for further details).
Since histamine-induced contractures (HICs) are also H1-mediatedin a rabbit saphenous vein (RSV), this led us to hypothesize that there might be other similarities between the action of carnosine and histamine in this tissue. The possible potentiating role of Zn ions in relation to actions of histamine itself have been presented in preliminary form [24] and are considered in a further paper [25].
In view of the likely relevance of Zn for the action of both histamine and carnosine, we have sought evidence that dynamic Zn release could occur near vascular smooth muscle. Since the mast cell is the acknowledged site for histamine release in the adventitial layer of vascular tissue, we made use of the suitably Zn-selective, cell impermeant fluorescent probe Newport Green 7990 in conjunction with confocal microscopy in isolated lengths of rabbit saphenous vein. The veins were opened along their long axis and placed, adventitial side uppermost, in a small chamber under the microscope objective.
Figure 5 shows the time course of Newport Green 7990 fluorescence detected in a region of apparently featureless extracellular space. The initial level shown upon including the dye is at least partly attributable to bathing [Ca2+] (0.2 mM). Upon bolus application of Compound 48/80, the selective mast cell degranulator, the extracellular fluorescence rose abruptly. A second “release” was provoked by a second application of 48/80. The divalent cation chelator EDTA was able to reduce the signal transiently (intensity was near saturation level at this sensitivity setting). The changes were mirrored in four regions of the same microscopic field.
In these circumstances, the dye is effectively sensitive only to ionized Zn; the Kd for Zn is ~1 µM, while those for Ca >1 mM, or Mg >10 mM, would make even quite large changes in the concentration of these ions virtually undetectable against their background levels. Direct calibration is not meaningful since the extracellular space is open to the bath volume (0.5 ml). Similar results were found in 8 preparations from 4 animals.Fig. 5. Time course of fluorescence of Newport Green 7990 (10 µM) (extracellular) from adventitial layer of a rabbit saphenous vein. The initial steady level after applying the dye (attributable partly to 0.2 mM calcium present) is sustained until Compound 48/80 is applied (10 µg/ml). On each application, a rapid, sustained rise in fluorescence is detected, indicating a rise in free Zn2+. The signal is reduced by a bolus injection of 20 µM EDTA (final concentration). See text for details.
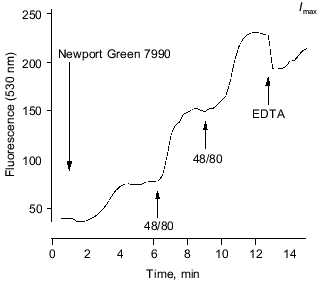
In the presence of 50 µM EDTA, the same maneuvers evoke no significant rise in fluorescence. This is consistent with the suggestion that the signal reports Zn ion release. If Ca release, for example, contributed significantly to the result illustrated, the observed fluorescence increase would represent a [Ca2+] rise of the order of 0.2 to 0.5 mM or more. However, such a large increase would hardly be affected and certainly not blocked, by just 50 µM EDTA. The concentration at the site of release from the mast cells would have to be higher still. EDTA binds Zn avidly under these conditions (despite competition from Mg and Ca ions, since the apparent affinity for Zn is several orders of magnitude higher) ensuring that free Zn remains well below 0.1 µM in the face of any plausible level of tissue Zn release. (Note that EDTA will not prevent degranulation by 48/80 here, where Ca remains present to excess). We believe these are the first reports using this dye and also the first of potentially significant Zn release in non-neuronal tissue.
DISCUSSION
We have demonstrated (Fig. 1) that the endogenous histidyl dipeptide carnosine can induce large sustained contractures (CICs) in vascular smooth muscle. Blockage by the divalent cation chelator EDTA and potentiation by adding Zn, but none of several other transition metals, strongly suggest that the effect is due to the formation of a Zn·Carn complex. Figure 2 shows that the results obtained with a very wide range of concentrations of Zn and carnosine can be reduced satisfactorily to a single curve if they are expressed in terms of the concentration of the complex. We can, therefore, confidently conclude that Zn·Carn is the active species and is effective with an apparent K0.5of 7..·10-8 M. However, there is a residual response to 10 mM carnosine in the presence of EDTA (even at 100 µM, data not shown) where Zn·Carn is calculated to be at least an order of magnitude lower than 10-8 M. A fraction of the response to carnosine is thus not easily attributable to interactions involving contaminating Zn ions, although carnosine might perhaps interact with tissue-bound Zn inaccessible to EDTA.
The relaxation effects of carnosine (with H1 actions blocked) are cimetidine-sensitive and thus probably H2-mediated. However, this effect is much less potent than that of histamine, significant relaxation occurring only at 1 mM carnosine or more (compared with 10 µM threshold for histamine). Neither carnosine- nor histamine-induced relaxation was potentiated by Zn. Nevertheless, these observations suggest that carnosine, like histamine, could have net hyper- or hypotensive actions, depending upon the balance between H1 or H2 receptors throughout the cardiovascular system. This helps to explain the hypotensive carnosine effect reported so long ago in vivo [10, 11]. Consistent with this conclusion, while histamine contracts many isolated vascular preparations, it is hypotensive in vivo, revealing dominant, H2-mediated relaxation [26]. Histamine-induced relaxation in arterial preparations seems to be via the endothelial H1 receptor [27], but we have yet to explore this effect in relation to carnosine. The cyclooxygenase inhibitor indomethacin, at a concentration that should inhibit prostaglandin synthesis, had no effect on the carnosine contracture but depressed that to histamine by a quarter. Qualitatively and quantitatively similar effects reported for isolated human saphenous vein [28] were attributed to indirect (H1-mediated) release of prostanoids from vascular endothelium. If carnosine acts via the same receptor as histamine, it is not clear why it did not elicit this response and rather implies that the two agents may not completely overlap in terms of receptor binding. Whether this implies a difference between smooth muscle and endothelial H1 receptors is open to speculation.
Schild analysis of mepyramine antagonism of the histamine and carnosine contractile responses yielded pA2 values of 8.47 and 7.97, respectively; reported pA2 values for mepyramine in venous preparations are generally between 8 and 9 [28, 29], with our values for histamine/mepyramine antagonism actually identical to those obtained by Schoeffter and Godfraind in human saphenous vein. In both cases, the slopes obtained are significantly different from unity indicating non-competitive antagonism. The details were discussed elsewhere [22] where we concluded they are consistent with the binding of histamine and carnosine at the same (i.e., H1) receptor. We were additionally able to confirm that histamine-induced contractures, like those evoked by carnosine, are very sensitive to methiothepin and moderately sensitive to ketanserin (unpublished observations) suggesting that the sensitivity of carnosine-induced contractures to these serotonergic antagonists was coincidental. Consistent with these findings, it has previously been reported that an azide derivative of ketanserin can irreversibly bind to the H1 receptor [30, 31] and cross sensitivity between 5-HT2 and H1 sites has also been noted for [3H]mianserin [32]. Alternatively, the possibility always remains that carnosine could be acting at a novel receptor with coincidental sensitivity to mepyramine and the other antagonists so far tested.
The action of carnosine acting via a vascular muscle receptor is thus virtually unique in two ways: it is a dipeptide agonist that is effective in the form of a metal chelate. The only other dipeptide action we know of is for N-acetylaspartylglutamate (NAAG) acting at the N-methyl-D-aspartate subclass of ionotropic acidic amino acid receptors [33]. We know of no other evidence for a metal chelate, as opposed to the ion itself (see below), acting through a receptor.
Physiological conditions for carnosine's actions. A circulating carnosinase ensures that general plasma concentrations of the dipeptide remain low [34]. Nothing is known about interstitial levels, local release, uptake, or synthesis in relation to vascular smooth muscle. In circumstances where extracellular carnosine concentration might rise, e.g., for muscle vasculature during or after exercise, or the mesenteric beds after carnosine-rich meat ingestion, concentrations achieved remain to be established. However, it must be borne in mind that because the maximum tensions evoked by Zn·Carn in vascular smooth muscle are so very large, even low levels of the conjugate could be physiologically relevant. Saphenous vein, which we have studied most, drains skeletal muscle in the hind limbs and might be exposed to higher concentrations of carnosine than some other vessels, but we found saphenous artery to behave in a very similar way. Carnosine induces contracture in several porcine vessels (also potentiated by Zn) (V. Wilson and colleagues, personal communication). Rat aorta, however, proved totally insensitive, even when Zn ions were added (data not shown).
In vivo competition with plasma albumin for Zn ions probably keeps circulating [Zn·Carn] near or below the threshold for vasoactivity (see Fig. 4 in [2]). However, Zn·Carn formation could be more significant in regions from which albumin is largely excluded (notably interstitial and cerebrospinal fluids).
A metabolic link between carnosine, carcinine (beta-alanyl-histamine), and histamine [35] has been established. Carnosine (via histidine) has been suggested to form as a non-mast cell source of histamine during physiological “shock” [36] in many histamine-rich tissues. The view of carnosineas a functionally inert histamine precursor must be re-evaluated especially where overlap of carnosine and H1 receptor availability is known [37]. The possible receptor-modulated physiological role for carnosine and its relationship with Zn was discussed further elsewhere [12].
It is worth noting that a novel anti-ulcer drug, Z-103, recently developed (Hamari Chemicals Ltd, Japan), is a stable complex of Zn and carnosine [38]1. However, pharmacological screening of the compound has indicated no antagonistic action at H1 or H2 receptors [39].
1 Editor's note: See also paper by Matsukura and Tanaka in this issue.
Our findings could well have significance for other tissues where both carnosine and Zn are present. The Zn·Carn complex could be formed significantly in brain where there are carnosine-rich regions and local Zn concentrations can reach 300 µM [8, 40]. Zn2+ can modulate neuronal activity [41] by affecting central neuron excitability (via glutamate and GABA receptors subtypes) and is released from active hippocampal neurons [40, 42]. Whilst a “co-transmitter” or “neuromodulator” role for carnosine in the central nervous system has been suggested, interaction with Zn has only been speculated upon [8]. Homocarnosine (GABA-L-histidine), a closely related dipeptide present in several brain regions, is only slightly vasoconstrictive in rabbit saphenous vein even when potentiated by Zn ions (data not shown).
Our novel results with the Zn-sensitive dye Newport Green 7990 (Fig. 5) provide new evidence that mast cells in the adventitia of blood vessels may release significant amounts of Zn when induced to degranulate. Since this occurs in the extravascular space, competition with albumin for these Zn ions is much reduced so that carnosine, or histamine itself, may have access to a functionally significant Zn reservoir. As a further aspect, Zn interaction with carnosine might affect the activity of Zn-containing metalloproteins such as angiotensin- and endothelin-converting enzymes (present in the interstitial space) and the Cu-Zn form of superoxide dismutase. Inhibitors of angiotensin converting enzyme (ACE), some of which act by a Zn binding [43] mechanism, are arguably the most significant recent development in the management of high blood pressure. Thus, our observations reveal a previously unsuspected endogenous agonist-receptor system whose relevance could extend beyond its direct effects on vascular smooth muscle.
In summary, our current understanding is that the vasoconstricting action of Zn·Carn in rabbit saphenous vein is mediated via an H1 (mepyramine-sensitive) receptor. In addition, carnosine has a less potent relaxing action, probably via an H2 (cimetidine-sensitive) receptor. This may help to explain carnosine's hypotensive action in vivo. Carnosine must be increasingly considered as a potent agonist whose full physiological significance is only slowly being appreciated.
We thank the British Heart Foundation and the Medical Research Council for financial support. Helpful discussion on various aspects of this work with our colleagues J. C. McGrath, C. Daly, and B. Morris and the Zn2+ analyses by the Chemistry Department of the University of Glasgow are gratefully acknowledged.
REFERENCES
1.Chapman, R A., and Miller, D. J. (1974) J.
Physiol., 242, 615-634.
2.Lamont, C., and Miller, D. J. (1992) J.
Physiol., 454, 421-434.
3.Crush, K. (1970) Comp. Biochem. Physiol.,
34, 3-30.
4.Miller, D. J., Lamont, C., and O'Dowd, J. J. (1993)
in Calcium Sensitising: a Novel Inotropic Mechanism, Chap. 5
(Allen, D. G., and Lee, J., eds.) Publ. Oxford University Press, pp.
117-139.
5.Dunnett, M., and Harris, R. C. (1992) J.
Chromatogr. Biomed. Appl., 579, 45-53.
6.Boldyrev, A. A., and Severin, S. E. (1990) Adv.
Enzyme Regul., 30, 175-194.
7.MacFarlane, N., McMurray, J., Dargie, H., O'Dowd,
J. J., and Miller, D. J. (1991) J. Molec. Cell. Cardiol.,
23, 1205-1207.
8.Sassoe-Pognetto, M., Cantino, D., and Fasolo, A.
(1992) Brain Res., 578, 261-268.
9.Harrison, S. M., Lamont, Ch., and Miller, D. J.
(1985) J. Physiol., 371, 197P.
10.Mason, E. C., and Binkley, S. (1931) Ann. Int.
Med., 4, 1319-1327.
11.Du Vigneaud, V., and Hunt, M. (1936) J. Biol.
Chem., 115, 93-100.
12.O'Dowd, A., O'Dowd, J. J., and Miller, D. J.
(1996) J. Physiol., 495, 535-543.
13.Daly, C. J., McGrath, J. C., and Wilson, V. G.
(1988) Brit. J. Pharmacol., 95, 473-484.
14.Mulvany, M. J., and Aalkjaer, C. (1990)
Physiol. Rev., 70, 921-926.
15.Smith, G. L., and Miller, D. J. (1985)
Biochim. Biophys. Acta, 839, 287-299.
16.Sillån, L. G., Martell, A. E., Hogfeldt, E.,
and Smith, R. M. (1974) Critical Stability Constants, Chem. Soc.
5. (London), Special publication No. 5 and supplements.
17.Pettit, L. D., and Powell, H. K. J. (1993)
International Union of Pure and Applied Chemistry Stability
Constants Database, Academic Software, Otley, UK.
18.Lentner, C. (ed.) (1981, 1984) Geigy
Scientific Tables, Vols. 1 and 3, Publ. Ciba Geigy (Basle).
19.Martin, G. R., and MacLennan, S. J. (1990)
Naunyn Schmiederberg's Archiv für Phamakologie, 342,
111-119.
20.Van Heuven-Nolsen, D., Tysse Klasen, T. H. M.,
Luo, Q., and Saxena, P. R. (1990) Eur. J. Pharmacol.,
191, 375-382.
21.Ash, A. S. F., and Schild, H. O. (1966) Brit.
J. Pharmacol. Chemother., 27, 427-439.
22.O'Dowd, A., and Miller, D. J. (1998) Brit. J.
Pharmacol., 125, 1272-1280.
23.Treherne, J. M., Stern, J. S., Flack, W. J., and
Young, J. M. (1991) Brit. J. Pharmacol., 103,
1745-1751.
24.O'Dowd, A., and Miller, D. J. (1998) J.
Physiol., 513P, 117P.
25.O'Dowd, A., and Miller, D. J. (2000),
submitted.
26.Powell, J. R., and Brody, M. J. (1976) J.
Pharmacol. Exp. Ther., 196, 1-14.
27.Van de Voorde, J., and Leusen, I. (1983) Eur.
J. Pharmacol., 87, 113-120.
28.Schoeffter, P., and Godfraind, T. (1989)
Pharmacol. Toxicol., 64, 64-71.
29.Tsuru, H., Iwata, M., and Shigei, T. (1983)
Experientia, 39, 577-578.
30.Wouters, W., van Dun, J., Leysen, J. E., and
Laduron, P. (1985) J. Biol. Chem., 260, 8423-8429.
31.Schotte, A., and Leysen, J. E. (1988) Eur. J.
Pharmacol., 145, 213-216.
32.Peroutka, S. J., and Snyder, S. H. (1981) J.
Pharmacol. Exp. Ther., 116, 142-148.
33.Valivullah, H. M., Lancaster, J., Sweetnam, P.
M., and Neale, J. H. (1994) J. Neurochem., 63,
1714-1719.
34.Gardner, M. L. G., Illingworth, K. M., Kelleher,
J., and Wood, D. J. (1991) J. Physiol., 439, 411-422.
35.Flancbaum, L., Brotman, D. N., Fitzpatrick, J.
C., van Es, T., Kasziba, E., and Fisher, H. (1990) Life Sci.,
47, 1587-1593.
36.Fitzpatrick, J. C., Fisher, H., and Flancbaum, L.
(1990) J. Surg. Res., 49, 293-297.
37.Hill, S. J. (1990) Pharmacol. Rev.,
42, 45-83.
38.Matsukura, T., Takahashi, T., Nishimura, Y.
Ohtani, T., Sawada, M., and Shibata, K. (1990) Chem. Pharm.
Bull., 38, 3140-3146.
39.Kurimoto, T., Jin, H., Takemasa, T., Sato, R.,
Ito, S., Furuichi, H., Yoshida, A., Ozeki, M., and Tagashira, E.
(1991) Pharmacometrics, 42, 69-82.
40.Assaf, S. Y., and Chung, S.-H. (1984)
Nature, 308, 734-736.
41.Smart, T. G., Xie, X., and Krishea, B. J. (1994)
Progr. Neurobiol., 42, 393-441.
42.Howell, G. A., Welch, M. G., and Frederickson, C.
J. (1984) Nature, 308, 736-738.
43.Zanchi, A., Nussberger, J., Criscuoli, M.,
Capone, P., and Brunner, H. R. (1994) J. Cardiovasc. Pharmacol.,
24, 317-322.