REVIEW: Carnosine and Protein Carbonyl Groups: A Possible Relationship
A. R. Hipkiss
Division of Biomolecular Sciences, GKT School of Biomedical Sciences, King's College London, Guy's Campus, London Bridge, London SE1 1UL, UK; fax: 0207-848-6399; E-mail: alan.hipkiss@kcl.ac.uk
Received November 1, 1999
Carnosine has been shown to react with low-molecular-weight aldehydes and ketones and has been proposed as a naturally occurring anti-glycating agent. It is suggested here that carnosine can also react with (“carnosinylate”) proteins bearing carbonyl groups, and evidence supporting this idea is presented. Accumulation of protein carbonyl groups is associated with cellular ageing resulting from the effects of reactive oxygen species, reducing sugars, and other reactive aldehydes and ketones. Carnosine has been shown to delay senescence and promote formation of a more juvenile phenotype in cultured human fibroblasts. It is speculated that carnosine may intracellularly suppress the deleterious effects of protein carbonyls by reacting with them to form protein-carbonyl-carnosine adducts, i.e., “carnosinylated” proteins. Various fates of the carnosinylated proteins are discussed including formation of inert lipofuscin and proteolysis via proteosome and RAGE activities. It is proposed that the anti-ageing and rejuvenating effects of carnosine are more readily explainable by its ability to react with protein carbonyls than its well-documented antioxidant activity.
KEY WORDS: carnosine, proteins, adducts, aging, aldehydes, methylglyoxal
My interest in carnosine was provoked by a letter from Robin Holliday who was working at CSIRO's laboratories in Sydney. He described some experiments, subsequently published in 1994 [1] and reconfirmed in 1999 [2], which showed that growth of cultured human fibroblasts with 20 or 30 mM carnosine delayed onset of cellular senescence and extended maximum cell division potential (the so-called Hayflick limit) by up to 10 doublings. Furthermore, addition of carnosine to senescent cells brought about a rejuvenating effect where the cells appeared to lose their senescent phenotype. These remarkable observations aroused my curiosity to the extent that I embarked upon a very enjoyable sabbatical year in Australia to try to get to grips with a possible mechanism that could explain carnosine's apparent anti-ageing actions. In this review I will describe a number of experiments that have led to the entirely novel proposal that carnosine directly participates in the metabolism of some senescent protein molecules.
At the time of my first acquaintance with carnosine (1990) the consensus of opinion was that carnosine was an anti-oxidant and free-radical scavenger [3-5]. However, other anti-oxidants and free-radical scavengers (e.g., vitamins C and E) did not have the same anti-ageing effects on the cultured human fibroblasts. This suggested that carnosine had important actions in addition to any anti-oxidant functions. The literature on carnosine even then was very diverse; other functions proposed included pH-buffering action, histidine source, transition metal ion chelator, neurotransmitter, wound healing agent, and immunostimulant [3]. I suspected that the mechanism of carnosine's apparent anti-ageing effects could involve any one or more of these proposed additional properties or even hither-to unrecognized activities [6]. I subsequently became aware that the suspicion that the main function of carnosine might be other than a direct anti-oxidant was shared by others [7]. Apart from the papers by McFarland and Holliday [1, 2], there have been few other studies of carnosine's effects on the lifespan of cultured cells. There has been one report that carnosine preserves the phenotype of rat fibroblasts when cultured at high oxygen tensions [8], and an earlier report that the dipeptide decreased chromosomal breakage [9]. Interestingly another dipeptide, beta-alanyl-cysteamine disulfide (beta-alethine) has also been reported to extend the Hayflick limit in cultured human fibroblasts [10]. That another beta-alanyl-peptide has an apparent anti-ageing action possibly points to the importance of the beta-peptide bond and the terminal amino group in modulation of the processes of ageing.
CARNOSINE AND CHEMICAL MODIFICATION TO PROTEINS
Previous workers on the “carnosine project” in Sydney had observed that protein cross-linking in human fibroblast cultures was depressed by carnosine. As luck would have it, immediately prior to my departure for Sydney I became interested in non-enzymic glycosylation of proteins, a process in which proteins are modified and eventually cross-linked by the action of reducing sugars [11]. I was able to suggest that the cross-links in the cultured fibroblasts might not result solely from the effects of reactive oxygen species (ROS) but could also derive from glycation, and perhaps carnosine modulated the latter process. This likelihood was reinforced when we realized that carnosine's structure resembled the preferred protein glycation sites, i.e., a target amino group with proximal imidazole and carboxyl groups [6, 12]. Our studies subsequently showed [13-15] that carnosine itself was a particularly good target for glycation by a variety of sugars and that the dipeptide could, sacrificially, protect other potential targets against glycation.
Non-enzymic glycosylation of proteins (glycation) is an age-associated phenomenon which is enhanced in uncontrolled diabetes and thought to be responsible for many of the secondary diabetic complications because of the formation of cross-links between modified (glycated) polypeptides and normal proteins [11]. Reactive aldehydes and ketones, and reducing sugars that cause glycation, have been implicated in many deleterious processes. For example, acetaldehyde and formaldehyde are normal but toxic metabolic products, and many glycolytic intermediates are also very effective glycating agents [16]. Our results show that carnosine can inhibit many aldehyde-mediated effects. For example, we found that the dipeptide could prevent formation of protein-protein cross-linking induced by deoxyribose [15], as well as protein-DNA cross-links induced by acetaldehyde and formaldehyde [17]. We showed that carnosine protected proteins against cross-linking induced by dihydroxyacetone, glyceraldehyde, and their phosphorylated derivatives [14, 15, 17]. We therefore proposed that carnosine might be a naturally occurring anti-glycating agent [6, 13-15, 17, 18]. That carnosine is more abundant in glycolytic (white muscle) than in more aerobic (red) muscle [8, 19] is consistent with this postulated protective function. The anti-glycating action of carnosine has also been confirmed by others [20-23] who also showed that the presence of the dipeptide could prevent aldehyde/sugar-mediated protein cross-linking and consequent enzymic inactivation.
The action of reactive oxygen species on lipids produces reactive end-products such as hydroxynonenal and malondialdehyde (MDA), both of which can modify proteins into forms characteristic of those usually associated with cellular and organism ageing [24]. We showed that carnosine could inhibit MDA-induced protein cross-linking as well as suppress formation of protein carbonyl groups [25], a modification that is particularly characteristic of ageing proteins and cells [26]. Presumably, carnosine also inhibits hydroxynonenal-mediated macromolecular modifications.
CARNOSINE AND METHYLGLYOXAL
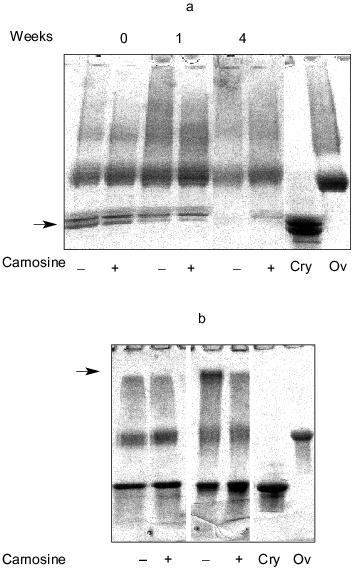
Methylglyoxal (MG) is another naturally occurring metabolite that can induce modifications to proteins that typify ageing [27]. Our results have shown that carnosine can also react with MG and prevent formation of MG-induced modification to proteins [28]. Furthermore, we recently found that carnosine can modulate the reactivity of the MG-treated protein [29]. When ovalbumin was treated with MG and then, following the removal of unreacted MG, incubated with alpha-crystallin with/without carnosine, we found that the presence of dipeptide inhibited cross-linking between the two polypeptides; the dipeptide inhibited both the disappearance of the crystallin as indicated by the arrow in Fig. 1a and the generation of slowly migrating material (presumably MG-ovalbumin cross-linked to alpha-crystallin) at the top of the gel as indicated by the arrow in Fig. 1b.
Fig. 1. The effects of carnosine on the cross-linking of methylglyoxal-treated ovalbumin (Ov) to alpha-crystallin (Cry). Ovalbumin was treated with 100 mM methylglyoxal (MG) for 2 weeks and then dialyzed exhaustively. The ability of the MG-treated protein to cross-link with alpha-crystallin added at 0.4 (a) or 2.5 mg/ml (b) was then determined by incubating the proteins together, with or without 50 mM carnosine, at 37°C for up to 4 weeks at pH 7 followed by polyacrylamide gel electrophoresis (PAGE) using Biorad 4-20% (Readygels). The arrows indicate the position of alpha-crystallin (a).
CARNOSINE MODULATES THE TOXICITY OF DELETERIOUS ALDEHYDES TO CULTURED CELLS
Carnosine can protect cells against the toxicity of aldehydes and ketones. For example, the dipeptide protected cultured human fibroblasts and lymphocytes against acetaldehyde [15]. Similarly, we found that carnosine protected cultured rat brain endothelial cells against MDA [30]. It is presumed that the first line of defense mediated by carnosine is the direct interaction with the toxic aldehyde, but other additional mechanisms are not ruled out such as sparing effects on cellular levels of the anti-oxidants glutathione (Dr. G. Grigg, personal communication) and vitamin E [31].
CARNOSINE AND THE REACTIVITY OF GLYCATED/OXIDIZED PROTEINS AND
AMINO ACIDS
The reaction of amino acids and proteins with reducing sugars and other aldehydes/ketones is to generate advanced glycosylation end-products (AGEs) [11]. These are produced via a process called the browning reaction or the Maillard reaction which is well known in food chemistry. Glycation products (AGEs) are generally deleterious and have also been termed glycotoxins [32]. Glycated proteins can be resistant to the attack by both extracellular and intracellular proteases and thereby decrease the availability of lysine, etc. [11]. Glycated proteins are immunogenic, being possibly responsible for the generation of auto-antigens associated with ageing [11]. Glycated proteins may also participate in the production of the protein cross-links also found in ageing and especially in the secondary complications and pathologies associated with diabetes; they can also provoke a hyperoxic response (the respiratory burst) in some cells. Furthermore, some glycated amino acids are mutagenic [33]. Interestingly, we found that glycated carnosine (i.e., when incubated with glucose) is not mutagenic, in contrast to the product of lysine's reaction with glucose, which is mutagenic [14].
The methods by which cells defend themselves against glycotoxins and AGEs are uncertain but can involve AGE interaction with scavenging receptors as well as specific AGE receptors (RAGEs) present on cells such as macrophages, fibroblasts, and endothelial cells. AGEs also bind to lysozyme, lactoferrin, and galectin [34]. However, as mentioned above, binding of AGEs to receptors can stimulate the generation of oxygen free-radicals (the respiratory burst), which can also be deleterious. It is possible that molecules such as carnosine might provide some sort of defense against the reactivity of certain glycation products, as well as inhibit their synthesis. We found that carnosine could inhibit the reaction of glycated lysine (lysine incubated with deoxyribose for 14 days) with protein [28]. Similarly, carnosine modulated the toxicity of such a product towards cultured cells [17]. Although we do not know the mechanism(s) by which carnosine's protection is mediated, direct reaction with the glycated moiety is one likely possibility.
EVIDENCE THAT CARNOSINE REACTS WITH AN “AGED”
PROTEIN
Protein ageing is associated with the accumulation of aberrant polypeptides, particularly those bearing carbonyl groups [26]. It is thought that carbonyl groups derive from (1) the direct oxidation of amino acid side chains by ROS, (2) the reaction of lipid peroxidation products MDA and hydroxynonenal with lysine, arginine, and histidine side chains, and (3) glycation phenomena where other deleterious aldehydes, including reducing sugars, induce formation of carbonyl groups in proteins via the Maillard reaction as well as other mechanisms.
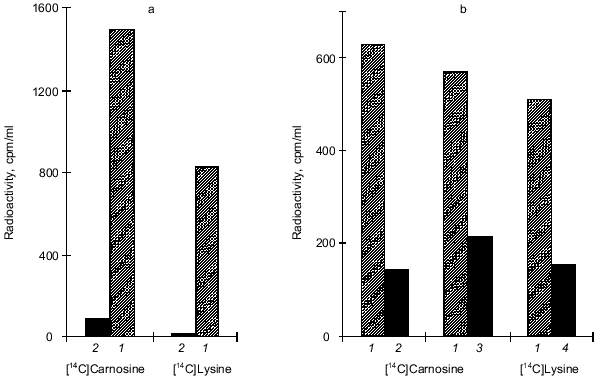
There is convincing evidence that carnosine can react with low-molecular-weight aldehyde/ketonic compounds [6, 13-15, 17, 20-22, 25, 28]. We therefore considered the possibility that carnosine could react with similar groups when they occur in polypeptide chains (i.e., protein carbonyls). Our most recent results suggest that carnosine can indeed react with protein carbonyl groups [29]. We modified ovalbumin with MG to induce formation of polypeptide carbonyl groups. Following this modification, the effect of subsequent incubation with carnosine on the reactivity of protein carbonyls was determined. Figure 2 shows that carnosine accelerated the loss of detectable carbonyl groups (as assayed by their reactivity with dinitrophenylhydrazine [35]). When radiolabeled carnosine was employed, we found that the modified protein became radiolabeled (Fig. 3). Such radiolabeling was inhibited by excess unlabeled lysine and N-acetyl-glycyl-lysine methyl ester, and conversely the attachment of radiolabeled lysine to the MG-treated protein was prevented by excess carnosine (Fig. 3). These results suggest that carnosine's amino group reacts with the protein carbonyl group to produce a protein-carbonyl-carnosine adduct. That carnosine not only reacts with protein carbonyl groups (Figs. 2 and 3) but also modulates their reactivity is suggested by the finding that the dipeptide inhibited formation of cross-links between the MG-treated protein and a normal unmodified polypeptide (alpha-crystallin) as shown in Fig. 1. We therefore propose the term “carnosinylation” for the attachment of carnosine to the protein carbonyl group, thus generating a “carnosinylated” protein (the protein-carbonyl-carnosine adduct).
Fig. 2. The effects of carnosine and lysine on the loss of carbonyl groups from methylglyoxal-treated ovalbumin. Ovalbumin was treated with 100 mM methylglyoxal (MG) for 2 weeks and then dialyzed exhaustively. The incubation was continued (pH 7, 37°C) either without additions (1) or in the presence of either 50 mM carnosine (3) or 50 mM lysine (2). Carbonyl groups were measured by their reactivity with dinitrophenylhydrazine [35] and expressed per mole of protein.
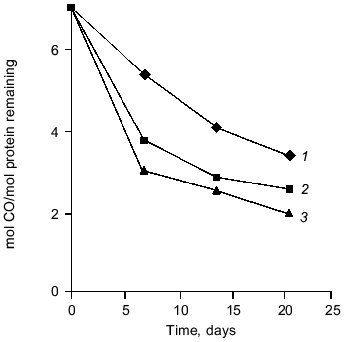
Fig. 3. a) The reaction of [14C]carnosine and [14C]lysine with methylglyoxal-treated (1) and untreated ovalbumin (2). Ovalbumin was treated with 100 mM methylglyoxal (MG) for 2 weeks, dialyzed to remove excess MG, and then incubated in the presence of either [14C]carnosine or [14C]lysine for 2 weeks. Then trichloroacetic acid (TCA) (5%)-precipitable radioactivity was determined. b) The effects of 50 mM lysine (2), 50 mM carnosine (4), and 50 mM alpha-acetyl-glycyl-lysine methyl ester (3) on the binding of radiolabeled carnosine and lysine to MG-treated ovalbumin(column 1, no addition). MG-treated ovalbumin (2 weeks) was dialyzed and then incubated (2 weeks) with either [14C]carnosine or [14C]lysine. Then TCA-precipitatable radioactivity was determined.
ARE “CARNOSINYLATED” PROTEINS PRODUCED IN CELLS?
At present we do not have any evidence that carnosine reacts with protein carbonyl groups intracellularly. Such studies are planned using radiolabeled carnosine and antibodies generated to the putative protein-carbonyl-carnosine adducts. However, should protein carnosinylation occur intracellularly, its occurrence might explain the controversy concerning the relative rates at which protein carbonyls accumulate during cell and organism ageing [36, 37]. Some workers suggest that protein carbonyls gradually accumulate during cell culture, whilst others find that protein carbonyls are found predominantly at the senescent stage. Variation in the relative extent of protein-protein and protein-carnosine cross-links together with age-related changes in carnosine synthesis/availability might help to provide some explanation for this dilemma.
THE POSSIBLE FATES OF “CARNOSINYLATED” PROTEINS
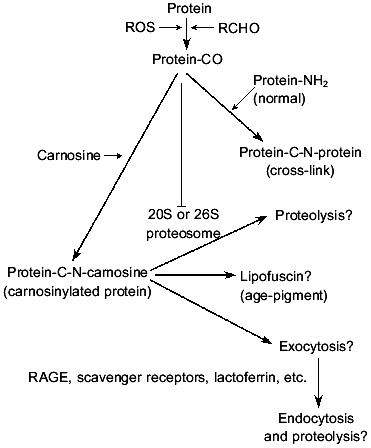
Our results suggest that the reactivity of the aged/glycated protein carbonyl groups is masked following reaction with carnosine. It is tempting to speculate on the possible fates of the postulated carnosinylated proteins (see Fig. 4). One possibility is that the carnosine adducts are a form of lipofuscin, the so-called age pigment. There has been some controversy about reactivity or toxicity of lipofuscin. Some workers have regarded it as an inert by-product [38], whereas others have claimed that it is deleterious [39] providing a major source of glycated/oxidized protein possessing reactive carbonyl groups. Certainly, lipofuscin is heterogeneous and hence the various claims for its cellular effects might be explained by the different lipofuscins employed. Perhaps the lipofuscin formed between protein/lipid carbonyls and carnosine is relatively inert, whereas the form generated in absence of carnosine would be capable of reacting with other cellular macromolecules via the carbonyl groups (see Fig. 1). Circumstantial support for carnosine's participation in lipofuscin synthesis is provided by the fact that lipofuscin is usually found in large amounts in tissues normally rich in carnosine (muscle, nerve, and brain).
Another possible fate of the carnosinylated proteins is proteolysis. Aberrant polypeptides are usually rapidly and selectively degraded, usually via the proteosome system, the 20S or 26S multicatalytic proteolytic particles [40]. Typical substrates include some oxidized proteins, but it should be noted that polypeptides subjected to high degrees of oxidative damage appear resistant to proteolytic attack and can even inhibit proteosome function [41] possibly via the action of carbonyl groups. The possibility arises therefore that carnosine might not only cap or mask carbonyl groups but also facilitate proteolysis of oxidized molecules. Ubiquitination of the carnosinylated proteins might be possible should the beta-amino group of the carnosine become available to provide a site for isopeptide bond formation with the ubiquitin terminal (glycine) carboxyl group [18].Fig. 4. Hypothetical scheme on the postulated relationship between carnosine and glycated/oxidized protein (protein-CO) and the possible fates of the resultant carnosinylated adduct. ROS, reactive oxygen species; RCHO, reactive aldehyde or reducing sugar; protein-CO, protein bearing carbonyl; , inhibition of proteosome function.
Few studies of the effects of carnosine on intracellular protein turnover have been carried out, but we found [15] that proteolysis of certain slowly turning-over proteins was stimulated in fibroblasts grown with 30 mM carnosine, especially as the cells approached their normal Hayflick limit, compared to cells cultured in the absence of the dipeptide. Clearly, these experiments need to be extended, but they are consistent with the idea that carnosinylated proteins are selectively degraded.
Another possibility is that carnosine modifies AGEs to either prevent their interaction with scavenging or AGE receptors (RAGEs), or alter the cellular response by modulating signal transduction and subsequent generation of ROS [34]. These ideas are readily testable as such experiments are relatively easy to perform.
CARNOSINE AND CARBONYL GROUPS GENERATED IN LIPIDS AND DNA
Proteins are not the only source of reactive carbonyl groups. For example, the ethanolamine of phospholipids can become oxidized to generate a carbonyl group [42]. Hence, it is possible that carnosine could modulate their subsequent deleterious effects (by forming carnosinylated lipids) in phospholipid membranes in a manner similar to our proposals for protein carbonyls.
DNA is another potential source of macromolecular carbonyl/aldehyde groups. Spontaneous depurination, together with depyrimidination, are the most common forms of spontaneous DNA damage and result in breakage of the glycosidic bond between the purine and carbon atom number 1 of the deoxyribose [43]. The same bond is cleaved by the specific enzymically mediated depurination/depyrimidination that occurs during base excision repair via the action of DNA base glycosylases (e.g., uracil DNA glycosylase) [44]. Both processes generate a deoxyribose moiety capable of behaving as a deleterious aldehyde such as glucose, but with far greater reactivity. The fate of the deoxyribose is uncertain, but some hypothetical schemes have suggested that a di-carbonyl moiety (4-oxo-2-pentenal) is eventually produced [45]; however, such schemes do not suggest possible fates for such highly reactive products. Hence, it is at least conceivable that carnosine could play a role in the disposal of the reactive deoxyribose, especially as the dipeptide has been shown to react with deoxyribose [14] and protect proteins against deoxyribose-mediated modification [14, 15], inhibit aldehyde-mediated DNA-protein cross-linking [17], and decrease chromosomal aberrations in cultured cells [9].
OTHER PROTECTIVE EFFECTS
Carnosine's ability to react with the hypochlorite anion is well documented [46] and most probably accounts for the dipeptide's protection of protein against hypochlorite-induced formation of carbonyl groups and cross-linking [25]. Carnosine as well as beta-alanine and homocarnosine have been shown to modulate the toxicity of the amyloid peptide fragment 25-35 towards cultured rat brain endothelial cells [47]. It is not understood how such protection is mediated, but carnosine has been reported to inhibit glycation-induced polymerization of the beta-amyloid peptide [48] into the presumably toxic multimeric form. Alternatively/additionally, oxidative events are also possible, and carnosine can react with MDA and other deleterious aldehydes generated subsequent to amyloid peptide cellular binding/entry [49]. It may also be relevant to note that beta-A4 peptide toxicity may be dependent on its interaction with the so-called ERAB protein, which may also possess low-molecular-weight alcohol dehydrogenase activity [50]. Consequently, if the protein does generate aldehydes from any low-molecular-weight alcohol, then one can propose that carnosine's aldehyde scavenging ability may play a role here as well. Another possibility is suggested by the observation that glucose utilization is markedly stimulated in cells exposed to the amyloid peptide together with carnosine [17, 47], which is consistent with the dipeptide facilitating some energy-driven detoxification process such as ATP-dependent proteolysis.
DO CELL-ASSOCIATED CARNOSINE CONCENTRATIONS CHANGE WITH
AGE?
It is possible that cellular concentrations of carnosine decrease with age (this is addressed in the review by H. J. Stuerenburg in this issue). If this is the case, then it is at least conceivable that in the young state carnosine concentrations are sufficient to permit carnosinylation of all potential sites. If carnosine levels are compromised for any reason during ageing, then availability of carnosine might become rate limiting for effective protein carnosinylation. It is perhaps interesting that amongst a range of mammals the muscular concentration of carnosine correlates with maximum lifespan of the species (1 mM in mice compared to 20 mM in humans) [6, 51, 52]. Hence, if carnosine levels in tissues do decrease gradually with age, then those species which commence life with the highest amount might reach the limiting value at much greater ages.
THE POSSIBLE MECHANISMS OF CARNOSINE'S ANTI-AGEING ACTIONS
The above speculations on carnosine's possible reaction with carbonyl groups present on proteins, lipids, and DNA degradation products can only partly explain its apparent anti-ageing action, i.e., reversion of the senescent phenotype [1, 2]. The proposal predicts (Fig. 4) that one should detect the transient attachment of carnosine to oxidized protein followed by either its degradation via the proteosome or accumulation as inert lipofuscin. Such ideas are testable. The proposal does not readily explain the effects of carnosine on the maximal cell division capacity (the Hayflick limit) of the cultured human fibroblasts [1, 2]. It has recently been shown however that telomere loss during cell division in cultured cells is a consequence of both the end-replication problem and ROS-mediated DNA damage, particularly as telomeric DNA does not appear to be repaired [53]. As most “standard” culture conditions (5% oxygen) are hyperoxic to the cells such as fibroblasts, then one could simply suggest that the ROS-scavenging properties of carnosine might contribute to an explanation why growth with dipeptide might decrease telomeric DNA damage and thereby permit extra cell divisions. However, one would predict that other anti-oxidants would have similar cellular effects, but this is apparently not the case. Hence, we suggest that the additional properties of carnosine that we propose here could play a role in the extension of the Hayflick limit particularly as it has been shown that oxidized polypeptide inhibits cell division, possibly as a result of compromised proteosome function [40, 41]. Perhaps the carbonyl binding activity of carnosine (or the released histidine) might mask any deleterious effects of the aberrant (carbonyl) protein on proteosome function as well as facilitate degradation (see Fig. 4) to allow the extras cell divisions. Indeed as ageing is multifactorial, then any anti-ageing agent such as carnosine should be pluripotent in its actions [6, 18]. However, it should be noted that should carnosine induce transcription of genes coding for proteins possessing homeostatic functions then this would provide additional/alternative explanations of carnosine's anti-ageing activity, especially should the proteins play a role in the removal of oxidized macromolecules. Indeed, it has recently been shown that carnosine does induce transcription of the vimentin gene as well as others [54]. Whether this is a direct specific effect of carnosine or merely related to the expression of proteins involved in the maintenance of cell structure is unsure. However, vimentin may be related to phagocytosis and both these parameters have been reported to be depressed with age in Kupffer cells [55]; this suggests the possibility that carnosine may regulate phagocytosis too.
THE CARNOSINASE PARADOX
Carnosine is one of the least toxic of compounds. Hence, the presence of intracellular and serum forms of the enzyme carnosinase that cleaves the molecule's peptide bond is a major paradox. Furthermore, congenital absence of this enzyme coupled with excretion of large amounts of carnosine is thought to be associated with mental retardation in humans [56]. It therefore follows that in some circumstances carnosine may have unrecognized properties that are deleterious in some way or that cleavage of the dipeptide is necessary for effective function. It is possible that the active molecule in the carbonyl/aldehyde binding in vivo is in fact histidine. Indeed, the reaction product formed between histidine and formaldehyde (spinacine) is found in human urine. Perhaps the natural toxicity of free histidine necessitates its storage as carnosine, and the presence of tissue and serum carnosinases liberate histidine only at specific locations and circumstances. If this is the case, then one would expect the attachment of histidine and not complete carnosine to protein carbonyl groups in the cell-associated scenarios described above. This should be reasonably easy to test using specific radiolabeling techniques and mass spectrometry.
Thus, carnosine may be a pluripotent protective (homeostatic) peptide; it can inhibit aldehyde/ketone-mediated modifications to protein and DNA, and can also modulate the reactivity of the aldehyde-modified protein by carnosinylation. Both levels of inhibition appear to occur by direct reaction between carnosine and the aldehyde/ketone/carbonyl group. It is possible, though not proven, that carnosinylation of protein carbonyls occurs intracellularly. Carnosinylated protein may either then be selectively degraded or provide a source of inert lipofuscin. These speculations should be regarded as additional to any other previously proposed homeostatic properties for this remarkable “simple” dipeptide.
REFERENCES
1.McFarland, G. A., and Holliday, R. (1994) Exp.
Cell Res., 212, 167-175.
2.McFarland, G. A., and Holliday, R. (1999) Exp.
Geront., 34, 35-45.
3.Quinn, P. R., Boldyrev, A. A., and Formazyuk, V. E.
(1992) Mol. Aspects Med., 13, 379-444.
4.Kohen, R., Yamamoto, Y., Cundy, K. C., and Ames, B.
N. (1988) Proc. Natl. Acad. Sci. USA,95, 2175-2179.
5.Aruoma, O. I., Laughton, M. J., and Halliwell, B.
(1989) Biochem. J., 264, 863-869.
6.Hipkiss, A. R., Holliday, R., McFarland, G., and
Michaelis, J. (1993) Lifespan, 4, 1-3.
7.Kansci, G., Genot, C., and Gandamer, G. (1994)
Science des Aliments, 14, 663-671.
8.Kantha, S. S., Wada, S., Tanaka, H., Takeuchi, M.,
Watabe, S., and Ochi, H. (1996) Biochem. Biophys. Res. Commun.,
223, 278-282.
9.Gille, J. J. P., Pasman, P., van Berkel, C. G. M.,
and Joenje, H. (1991) Mutagenesis, 6, 313-318.
10.Knight, G. D., Mann, P. L., Laubscher, K. H., and
Scallen, T. J. (1994) Cancer Res., 54, 5636-5642.
11.Baynes, J. W., and Monnier, V. M. (1989) The
Maillard Reaction in Aging, Diabetes and Medicine, Alan R. Liss, N.
Y.
12.Shilton, B. H., and Walton, D. J. (1991) J.
Biol. Chem., 266, 863-869.
13.Hipkiss, A. R., Michaelis, J., Syrris, P., Kumar,
S., and Lam, Y. (1994) Biochem. Soc. Trans., 22,
399S.
14.Hipkiss, A. R., Michaelis, J., and Syrris, P.
(1995) FEBS Lett., 371, 81-85.
15.Hipkiss, A. R., Michaelis, J., Syrris, P., and
Dreimanis, M. (1995) Perspect. Hum. Biol., 1, 59-70.
16.Hamada, Y., Araki, N., Koh, N., Nakamura, J.,
Homuchi, S., and Hotta, N. (1996) Biochem. Biophys. Res.
Commun., 228, 539-543.
17.Hipkiss, A. R., Preston, J. E., Himsworth, D. T.
M., Worthington, V. C., Keown, M., Michaelis, J., Lawrence, J., Mateen,
A., Allende, L., Eagles, P. A. M., and Abbott, J. (1998) Ann. N. Y.
Acad. Sci., 854, 37-53.
18.Hipkiss, A. R. (1998) Int. J. Biochem. Cell
Biol., 30, 863-868.
19.Kantha, S. S., Wada, S., Tanaka, H., Takeuchi,
M., Watabe, S., and Ochi, H. (1996) Biochem. Biophys. Res.
Commun., 223, 278-282.
20.Lee, B. J., Kang, K. S., Nam, S. Y., Park, J. H.,
Lee, Y. S., Yun, Y. W., and Cho, M. H. (1999) Korean J. Physiol.
Pharmacol., 3, 251-261.
21.Swearengin, T. A., Fitzgerald, C., and Seidler,
N. W. (1999) Molec. Toxicol., 73, 307-309.
22.Vinson, J. A., and Howard, T. B. (1996)
Nutritional Biochem., 7, 659-663.
23.Kuleva, N. V., and Kovalenko, Z. S. (1997)
Biochemistry (Moscow), 62, 1119-1123.
24.Miyata, T., Inagi, R., Asahi, K., Yamada, Y.,
Horie, K., Uchida, K., and Kurokawa, K. (1998) FEBS Lett.,
437, 24-28.
25.Hipkiss, A. R., Worthington, V. C., Himsworth, D.
T. M., and Herwig, W. (1997) Biochim. Biophys. Acta,
1380, 46-54.
26.Stadtman, E. R. (1992) Science,
257, 1220-1224.
27.Chaplen, F. W. R. (1998) Cytotechnology,
26, 173-183.
28.Hipkiss, A. R., and Chana, H. (1998) Biochem.
Biophys. Res. Commun., 248, 28-32.
29.Brownson, C., and Hipkiss, A. R. (2000) Free
Rad. Biol. Med., submitted.
30.Hipkiss, A. R., Preston, J. E., Himsworth, D. T.
M., Worthington, V. C., and Abbot, N. J. (1997) Neurosci. Lett.,
238, 135-138.
31.Chan, W. K., Decker, E. A., Chow, C. K., and
Boissoneault, G. A. (1994) Lipids, 29, 461-466.
32.Koschinski, T., He, C.-J., Mitsuhashi, T.,
Bucala, R., Liu, C., Buenting, C., Heitman, K., and Vlassara, H. (1997)
Proc. Natl. Acad. Sci. USA, 94, 6474-6479.
33.Kim, S. B., Kim, I. S., Yeum, D. M., and Park, Y.
H. (1991) Mut. Res., 254, 65-69.
34.Thornalley, P. J. (1998) Cell Mol. Biol.,
44, 1013-1023.
35.Uchida, K., Kanematsu, M., Sakai, K., Matsuda,
T., Hattori, N., Mizuno, Y., Suzuki, D., Miyata, T., Noguchi, N., Niki,
E., and Osawa, T. (1998) Proc. Natl. Acad. Sci. USA, 95,
4882-4887.
36.Goto, S., and Nakamura, A. (1997) Age,
20, 81-89.
37.Goto, S., Nakamura, A., Radak, Z., Nakamoto, H.,
Takahashi, R., Yasuda, K., Sakurai, Y., and Ischii, N. (1999) Mech.
Aging Devel., 107, 245-253.
38.Yin, D. Z. (1995) Gerontology, 41,
59-170.
39.Brunk, U. T., Jones, C. B., and Sohal, R. S.
(1992) Mut. Res., 275, 395-404.
40.Reinheckel, T., Sitte, N., Ullrich, O.,
Kuckelkorn, U., Davies, K. J. A., and Grune, T. (1998) Biochem.
J., 335, 637-642.
41.Sitte, N., Merker, K., and Grune, T. (1998)
FEBS Lett., 440, 399-402.
42.Bierhaus, A., Hofman, M., Ziegler, R., and
Nawroth, P. P. (1998) Cardiovasc. Res., 37, 586-600.
43.Lindahl, T. (1993) Nature, 362,
709-715.
44.Sancar, A. (1995) J. Biol. Chem.,
270, 15915-15918.
45.Nash, H. M., Bruner, S. D., Scharer, O. D.,
Kawate, T., Addona, T. A., Sponner, E., Lane, W. S., and Verdine, G. L.
(1996) Curr. Biol., 6, 968-980.
46.Boldyrev, A. A., Formazyuk, V. E., and Sergienko,
V. I. (1994) Sov. Sci. Rev. D. Physicochem. Biol., 13,
1-60.
47.Preston, J. E., Hipkiss, A. R., Himsworth, D. J.
T., Romero, I. A., and Abbott, N. J. (1998) Neurosci. Lett.,
242, 105-108.
48.Munch, G., Mayer, S., Michaelis, J., Hipkiss, A.
R., Schinzel, R. Riederer, P., and Cunninham, A. M. (1997) Biochim.
Biophys. Acta, 1360, 17-29.
49.Smith, M. A., Perry, G., Richey, P. L., Sayre, L.
M., Anderson, V. E., Beal, M. F., and Kowall, N. (1996) Nature,
382, 120-121.
50.Yan, S. D., Fu, J., Soto, C., Chen, X., Zhu, H.
J., AlMohanna, F., Collinson, K., Zhu, A. P., Stern, E., Saido, T.,
Tohyama, M., Ogawa, S., Roher, A., and Stern, D. (1997) Nature,
389, 689-695.
51.R. Holliday, personal communication.
52.Munch, G., Thome, J., Foley, P., Schinzel, R.,
and Riederer, P. (1997) Brain Res. Revs., 23,
134-143.
53.Von Zglinicki, T. (1998) Ann. N. Y. Acad.
Sci., 854, 318-327.
54.Ikeda, D., Wada, S., Yoneda, C., Abe, H., and
Watabe, S. (1999) Cell Struct. Function, 24, 79-87.
55.Sun, W. B., Han, B. L., Peng, Z. M., Li, K., Ji,
O., Chen, J., Wang, H. Z., and Ma, R. L. (1998) World J.
Gastroenterol., 4, 70-77.
56.Kunze, H., Kleinkauf, H., and Bauer, K. (1986)
Eur. J. Biochem., 160, 605-613.