Probing Contacts of Phosphate Groups of Oligonucleotides from the Non-Template Strand of lac UV5 Promoter with E. coli RNA Polymerase Using Regioselective Cross-Linking
E. A. Rudakova1*, M. G. Ivanovskaya1, M. V. Kozlov1, M. V. Khoretonenko1, T. S. Oretskaya1, and V. G. Nikiforov2
1Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (7-095) 939-3181; E-mail: erudakova@bioorg.chem.msu.ru2Institute of Molecular Genetics, Russian Academy of Sciences, pl. Kurchatova, Moscow, 123182 Russia; fax: (7-095) 196-0221; E-mail: vnikifor@img.ras.ru
* To whom correspondence should be addressed.
Received January 14, 2000; Revision received March 10, 2000
Contacts of phosphate groups at positions -12, -15, and -18 in relation to the transcription initiation site in the non-template strand of lac UV5 promoter with lysines or histidines of E. coli RNA polymerase in the open complex model were studied. A number of synthetic oligonucleotides from the -10-area of the non-template strand containing activated 5´-terminal phosphate group were cross-linked with holo- or core-enzyme of RNA polymerase. 5´-N-Hydroxybenzotriazole phosphodiesters of oligonucleotides were used as phosphate activated derivatives. They are capable of phosphorylating amino groups of lysines and histidines in the enzyme molecule that are brought into proximity with activated phosphate in the complex, resulting in the formation of a covalent bond between the oligonucleotide and the protein. The analysis of the products of cross-linking allowed the protein subunit and the amino acid residue taking part in the formation of the covalent bond for each oligonucleotide to be identified. It was found that all oligonucleotides from the non-template strand of promoter in the complex with the holo-enzyme are bound with the sigma70-subunit. When analyzing the products of partial cleavage of the complexes cross-linked at cysteines and methionines using SDS-PAGE, it was shown that phosphate at position -12 made contacts with His180 or His242 of the sigma70-subunit, the reactive amino acid residue being located between the first and second conservative regions. Phosphate at position -15 is located near lysines from two different areas--between Met413 and Met456 (regions 2.3 and 2.4) and between Met470 and Met507 (region 3.1). Phosphate at position -18 makes preferential contacts with a lysine situated between Met470 and Met507 (region 3.1). Based on the analysis of contacts of phosphate groups and the structure of the isolated sigma70-subunit established previously, a scheme of the mutual arrangement of the oligonucleotide and the sigma70-subunit possessed by the holo-enzyme has been proposed.
KEY WORDS: RNA polymerase from E. coli, oligonucleotide, cross-linking, activated phosphate group, lac UV5 promoter
Abbreviations: EDC) 1-ethyl-3-(3´-dimethylaminopropyl)-carbodiimide; HEPES) 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid; MES) 2-(N-morpholino)-ethanesulfonic acid; NHBT ester) N-hydroxybenzotriazole phosphodiester; 1-MeIm) 1-methyl imidazole.
All genes of the bacterium Escherichia coli are transcribed by
DNA-dependent RNA polymerase. The enzyme consists of five subunits,
four subunits (alpha2betabeta´)
forming the core-enzyme that performs processive elongation of a
nascent RNA chain. Attachment of the sigma-subunit to the
core-enzyme provides recognition of promoter and initiation of
transcription, the responsibility for transcription of the majority of
genes being on the sigma70-subunit [1]. Such a whole enzyme (holo-enzyme) has the ability
to recognize certain specific sequences of DNA-promoter located in the
regions -35 and -10 in relation to the transcription initiation site,
resulting in the formation of originally closed promoter complex [2]. Then a local melting of DNA-promoter strands in
the region adjacent to the transcription initiation site occurs with
the formation of the open promoter complex. At this stage, RNA
polymerase is capable of starting synthesis of RNA in the presence of
ribonucleotides [3]. The structure of the open
complex has been the subject of much investigation. Therefore, it was
shown that at this stage of recognition contacts between the
non-template strand of the promoter and holo-enzyme are primarily
formed. Results of experiments conducted using luminescence, point
mutagenesis, and covalent cross-linking of RNA polymerase with promoter
suggested that the -10 region of non-template strand of promoter
interacts primarily with C-terminal conservative region 2 of the
sigma70-subunit in the holo-enzyme [3-6].
It is known that the enzyme is capable of forming high-affinity specific complexes with short single-strand fragments of DNA that are analogs of the non-template strand of promoter containing the consensus TATAAT-sequence in the -10 region [7-9]. A number of methods were used to study the structure of the complex of RNA polymerase with oligonucleotides [10, 11]. Among these methods, cross-linking in the native complex is of particular interest because this method characterizes most adequately the point contacts in the protein-DNA (oligonucleotide) complex in solution. It was shown recently that when the complex of RNA polymerase with a synthetic 19-membered oligonucleotide (a fragment of the -10 region of the non-template strand of lac UV5 promoter) was exposed to UV light, a covalent bond was formed between the oligonucleotide and region 2 of the sigma70-subunit, i.e., the region interacting with the -10 region of the whole promoter [12]. It is worth noting that binding is strictly specific, i.e., the oligonucleotide that does not contain the consensus sequence corresponding to the -10 region of the promoter is unable to form a covalent bond with RNA polymerase [12]. This result suggests that interactions in such a model complex simulate the natural interactions in the open promoter complex, and the approach based on using short synthetic fragments of promoter gives an adequate notion of the structure of the RNA polymerase-DNA complex.
The goal of the present work was to probe the contacts of phosphate groups of synthetic oligonucleotides of different length from the -10 region of the non-template strand of promoter lac UV5 with the certain regions of E. coli RNA polymerase in the open complex model. Previously, the contacts of the ribose-phosphate backbone of the lac UV5 promoter with individual subunits of RNA polymerase in holo- and core-enzymes were determined [13, 14]. For this purpose, partial depurinization of the DNA promoter was carried out after its methylation by dimethyl sulfate. Then the positions of lysines of RNA polymerase bound covalently with aldehyde groups of depurinized regions of DNA were determined. It is evident that such data characterize the interactions between the enzyme and the modified promoter. It is worth noting that charged alkylated regions in DNA can strongly distort the structure of the native complex with the enzyme.
We tested the contacts of “zero length” between 5´-phosphate of a series of 12-22-membered oligonucleotides in the -10 region of template or non-template strand of lac UV5 promoter and nucleophilic groups of amino acid residues of the protein. Such contacts are of electrostatic nature and play a stabilizing role in the DNA-protein complex. To solve the problem, we cross-linked oligonucleotides with RNA polymerase in the native complex using N-hydroxybenzotriazole phosphodiesters (NHBT esters) of oligonucleotides. If the NHBT-activated phosphate group of the oligonucleotide and nucleophilic groups of lysines and histidines of the protein are brought into proximity in the complex, phosphorylation of the amino group of lysine or the imidazole group of histidine occurs easily, with formation of a covalent bond between protein and DNA [15]. Therefore, the results of cross-linking give an indication of the contacts of phosphate groups of DNA with nucleophilic groups of lysines and histidines of the protein in the studied complex.
When bound with the enzyme, the oligonucleotide can interact with one or several subunits of the RNA polymerase. The products of covalent binding with different subunits may be discriminated by SDS-PAGE. The efficiency of covalent binding of activated oligonucleotides with individual subunits in the complex containing holo- or core-enzyme was determined by radioautography after electrophoretic separation of the products of the reaction using SDS-PAGE. The total yield of the products of cross-linking varied from 0.5 to 40% as the contact phosphate-Lys (His) becomes stronger.
The difference in the resistance of the phosphoamide bond of phospholysine or phosphohistidine derivative to hydrolysis was used to identify the amino acid residue (lysine or histidine) participating in the covalent binding with oligonucleotide.
To determine the region of a subunit containing lysine or histidine cross-linked with oligonucleotide, the cross-linked complex was cleaved by BrCN or 2-nitro-5-thiocyanbenzoic acid at methionine or cysteine, respectively, under circumstances where no more than one break per protein molecule is formed [16]. The masses of the cleavage products determined by SDS-PAGE were compared with the masses of all the fragments formed in the total cleavage of a given subunit at methionines or cysteines. This procedure allows the regions of attachment of oligonucleotide to a subunit to be determined with a precision within the limits of the distances between neighboring methionines and cysteines [16].
Based on the data obtained for different oligonucleotides from the -10 region of the non-template strand of the promoter, the contacts of phosphate groups (at positions -12, -15, and -18 in relation to the transcription initiation point) with the sigma70-subunit possessed by holo-enzyme were determined. Taking into account the data of X-ray studies of isolated sigma70-subunit, a scheme for the mutual arrangement of oligonucleotide and sigma70-subunit in the cross-linked complex is proposed.
The high efficiency of cross-linking can be used for design of drugs that are capable of interacting irreversibly with RNA polymerase and act as regulators of transcription.
MATERIALS AND METHODS
Oligonucleotides were synthesized in the Laboratory of Nucleic Acid Chemistry of the School of Chemistry of Lomonosov Moscow State University by the amidophosphite method using an Applied Biosystems 380 B synthesizer (USA). Core-enzyme of RNA polymerase was isolated as described in [17] with some changes involving HPLC on Superose 6 (10/30 HR, Pharmacia Biotech, Sweden) and subsequent ion-exchange chromatography on Mono Q 5/5. The sigma70-subunit was isolated as described in [18].
The following reagents and enzymes were used in the present paper: propylene diamine, 1-methyl imidazole (1-MeIm), 1-ethyl-3-(3´-dimethylaminopropyl)-carbodiimide (EDC), 2-(N-morpholino)-ethanesulfonic acid (MES), MgCl2, and NaCl from Merck (Germany); Tris, EDTA, LiClO4, and (NH4)2S2O8 from Fluka (Switzerland); acrylamide, N,N´-methylene bisacrylamide, HEPES, and glycerol from Serva (Germany); SDS, N,N,N´,N´-tetramethylethylene diamine, and bromophenol blue from Reanal (Hungary); [gamma-32P]ATP (1000 Ci/mmole) from Izotop (Russia); and polynucleotide kinase T4 (25,000 U/ml) from SibEnzym (Russia).
Gel electrophoresis was carried out in 20%-polyacrylamide gels containing 7 M urea (0.05 M Tris, 0.05 M H3BO3, 1 mM EDTA, pH 8.3). Substances being analyzed were dissolved in 80%-formamide containing 0.01% bromophenol blue and 1 mM EDTA and then placed on the gel. Oligonucleotides were extracted from the gel by incubation in 2 M aqueous solution of LiClO4 for 12 h at 4°C.
32P was introduced in oligonucleotides using polynucleotide kinase T4 in the presence of [gamma-32P]ATP [19]. The efficiency of the reactions of oligonucleotides with protein or amine was estimated by measurement of radioactivity of the corresponding areas of the gel using the Cherenkov method of counting in a Tracor scintillation counter (The Netherlands).
Synthesis of NHBT esters of oligonucleotides. 5´-32P-phosphorylated oligonucleotides were treated with 2 M N-hydroxybenzotriazole in the mixture water-dimethyl formamide (1:3 v/v), pH 3.5, in the presence of 0.5 M EDC [15]. To estimate the degree of conversion of oligonucleotide, 10% of the substance formed was dissolved in 5 µl of 0.5 M aqueous propylene diamine, pH 8.0, and incubated for 2 h at 37°C. The efficiency of the formation of active NHBT esters was assessed by the accumulation of amino propylamide of oligonucleotide [15]. The yield of amino propylamides of oligonucleotides was 85-95%.
Noncovalent binding. Reaction mixtures containing 2 pmoles of protein (holo- or core-enzyme) and 0.2 pmole of 32P-labeled oligonucleotide in 10 µl of buffer (50 mM HEPES, pH 8.0, 40 mM NaCl, 5 mM MgCl2, 5% glycerol) were incubated for 15 min at 18°C. After addition of 2 µl of 0.01% solution of bromophenol blue in glycerol, the mixtures were placed in polyacrylamide gels precooled to 7°C under non-denaturing conditions. The composition of the polyacrylamide gel was the following: 4% acrylamide, 0.15% N,N´-methylene bisacrylamide, 0.09 M Tris, 0.08 M H3BO3. The positions of 32P-labeled compounds corresponding to non-bound oligonucleotide and oligonucleotide bound to protein were determined by radioautography.
Covalent binding. In the case of analytical cross-linking, the reaction mixtures (total volume 15 µl) contained 0.2-1 pmole of NHBT ester of 32P-labeled oligonucleotide and 1-8 pmoles of protein (holo- or core-enzyme) in the transcription buffer (20 mM HEPES, pH 7.8, 50 mM NaCl, 10 mM MgCl2). In the case of preparative cross-linking, we used 5-15 pmoles of NHBT ester of oligonucleotide and 15-30 pmoles of protein. The mixtures were incubated for 15-120 min at 37°C. Then 5 µl of the solution used for placing on polyacrylamide gel (0.5 M HEPES, pH 8.0, 30% glycerol, 3% SDS, 0.02% beta-mercaptoethanol, 0.1% bromophenol blue) were added to the reaction mixtures. To destroy the noncovalent complexes of protein with RNA, the samples were incubated for 7 min at 90°C and placed in 8% SDS gels (to separate sigma70, beta + beta´, and alpha-subunits) or 4% SDS gels (to separate beta- and beta´-subunits). The positions of 32P-labeled compounds were determined by radioautography.
The amino acid residue covalently bound with oligonucleotide was identified by cleavage of the covalent bond between protein and DNA. The cross-linked complex was isolated from the gel by elution in 500 µl of 0.5% SDS for 1 h and dried in vacuum by freezing in a SpeedVac centrifuge. Then the complex was analyzed by treatment by 0.01 M HCl, 0.05 M MES, pH 5.0, or 0.05 M 1-MeIm, pH 8.0, for 10-240 min at 37°C.
The samples were placed in 4% or 8% SDS gels and electrophoresis was carried out as described above. The positions of 32P-labeled areas were determined by radioautography.
Determination of the region of subunit bound to oligonucleotide. The subunit covalently bound with oligonucleotide was isolated from the gel by elution in SDS as described above. To cleave at methionines, the precipitate was dissolved in 30 µl of 0.05 M HCl. To determine the “zero point”, a third of the solution was frozen, and 1 µl of 1 M solution of BrCN in acetonitrile was added to the remainder of the solution [16]. Within 5 and 10 min the reaction proceeding at 18°C was terminated by addition of 5 µl of the solution used for placing the sample on the gel (the composition was given above). To cleave at cysteines, the precipitate was dissolved in 30 µl of 0.2 M Tris-buffer, pH 8.45, containing 7 M urea and 5 mM beta-mercaptoethanol. After incubation for 15 min at 37°C, one third of the solution was frozen to determine the “zero point”; 3 µl of 0.5 M solution of 2-nitro-5-thiocyanbenzoic acid in methanol was added to the remainder of the solution. After additional incubation for 15 min at 37°C, the pH of the reaction mixture was adjusted to 9.0 by addition of 1 M NaOH. Cleavage was carried out for 10 and 40 min at 37°C. The reaction was terminated by addition of 1 µl of 1 M HCl, 5 µl of solution used for placing on gel, and 1.7 µl of 10%-solution of SDS. The products of cleavage of the complex cross-linked at methionines and cysteines were separated in gradient (8-16%) or homogenous (8%) SDS gels.
RESULTS AND DISCUSSION
The following synthetic oligonucleotides from the -10 region of the lac UV5 promoter was used in the present work:

As a control oligonucleotide we used (N)19 of statistical composition, i.e., oligonucleotide produced in such a way that each stage of condensation contained the whole set of syntons (A, T, G, C).
The 32P-labeled phosphate group was incorporated in the 5´-terminus of each oligonucleotide to prove that it is precisely this phosphate group that participates in the contact with RNA polymerase in the cross-linked complex. The bracket refers to the length of the oligonucleotide. The positions of phosphate under testing in relation to the transcription initiation site (-12, -15, -18, +4) on the 5´-terminus of the above oligonucleotides are italicized.
1. Formation of complex between RNA polymerase and oligonucleotides of different primary structure. Before cross-linking of oligonucleotides with RNA polymerase, we studied the ability of the above-mentioned oligonucleotides and the control oligonucleotide (N)19 of statistical composition to form complexes with holo- and core-enzymes of RNA polymerase.
For this purpose, the mixture of oligonucleotide (20 nM) and RNA polymerase (200 nM) was incubated under conditions indicated in the legend to Fig. 1. This figure shows the radioautograph of a 4% gel under non-denaturing conditions used for the registration of the complex formation. When analyzing the data presented in Fig. 1, we established that “16”, “19”, and “22” (oligonucleotides from the non-template strand of promoter containing TATAAT-block and the adjacent region extending up to the transcription initiation site) efficiently interact with the holo-enzyme. Oligonucleotide “12” from the non-template strand devoid the region after the TATAAT-block, oligonucleotide (N)19 of statistical composition, and oligonucleotide “22” from the template strand of the promoter showed practically no interaction with the holo-enzyme. The lack of binding of the oligonucleotide from the template strand and the oligonucleotide of statistical composition is consistent with the view that the holo-enzyme recognizes the sequence TATAAT in the oligonucleotide. However, judging from the lack of binding of “12”, the region extending after TATAAT in the direction toward the transcription initiation site is also essential to binding.
Complexing of oligonucleotides with core-enzyme is characterized by the same affinity for all oligonucleotides. This fact indicates that binding with core-enzyme is not sequence specific.Fig. 1. Formation of complexes between holoenzyme and oligonucleotides of different primary structure. Lane 1 corresponds to oligonucleotide “16”. Lanes 2-7 correspond to the reaction mixtures obtained by incubation of holo-enzyme with (N)19, “12”, “16”, “19”, “22”, and template “22”, respectively, for 15 min at 18°C (50 mM HEPES, pH 8.0, containing 40 mM NaCl, 5 mM MgCl2, and 5% glycerol). Separation was carried out in a 4% gel under non-denaturing conditions. 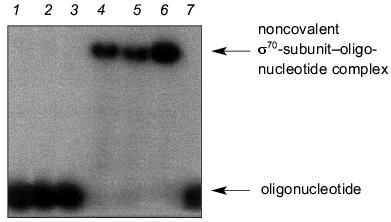
2. Covalent binding of oligonucleotides to holo- or core-enzyme of RNA polymerase. To determine what regions of RNA polymerase are responsible for binding with DNA, we studied covalent binding of the above oligonucleotides with holo- and core-enzymes under the conditions of existence of the native complex. The 5´-terminal phosphate group in all oligonucleotides selected was activated by the formation of NHBT ester. The activated oligonucleotide was incubated with RNA polymerase. To obtain NHBT esters, oligonucleotide was condensed with 2 M N-hydroxybenzotriazole using 0.5 M EDC [15]. The yield of NHBT esters was 85-90%. NHBT esters used for covalent binding with RNA polymerase are given below. The right upper index at the activated phosphate refers to its position in relation to the transcription initiation site.

NHBT esters of oligonucleotides are rather stable in water solutions at neutral pH. However, they are capable of phosphorylating strong nucleophiles with the formation of a covalent nucleophile-phosphate bond. Complexing of NHBT esters of nucleotides with a protein is accompanied primarily by the formation of covalent bonds between activated phosphate of the oligonucleotide and nucleophilic groups of lysines or histidines of the protein molecule. The yield of the product of cross-linking depends on the proximity of activated phosphate and the nucleophilic group of the protein.
Covalent binding of an NHBT ester of a oligonucleotide with holo- or core-enzyme was carried out by addition of the solution of oligonucleotide to the protein solution in the transcription buffer and incubation for 10-120 min at 37°C. Figure 2 shows the conditions and results of cross-linking. The reaction mixtures were separated by electrophoresis in 8% polyacrylamide gels during 60-150 min. Prolonged separation time was used on fine separation of products of covalent binding (Fig. 2c).
The results of separation of the reaction mixtures of “NHBT-16” and holo-enzyme after incubation for 10, 30, and 60 min in the transcription buffer are presented in Fig. 2a. The efficiency of covalent binding of “NHBT-16” with RNA polymerase reaches 25% within 1 h. It is not expedient to prolong the time of incubation, since this does not increase the yield of the products of cross-linking owing to partial hydrolysis of NHBT ester of the oligonucleotide.Fig. 2. Covalent binding of NHBT esters of oligonucleotides with holo-enzyme. The conditions of cross-linking are as follows: 20-100 nM DNA, 100-800 nM protein, 20 mM HEPES, pH 7.8, 50 mM NaCl, 10 mM MgCl2, 37°C. a) Accumulation of the products of covalent binding of “NHBT-16” with holo-enzyme. Time of incubation (min): 1) 0; 2) 10; 3) 30; 4) 60. b) Covalent binding of NHBT esters of oligonucleotides of different primary structure with holo-enzyme for 1-h incubation: 1) “NHBT-12”; 2) “NHBT-16”; 3) “NHBT-19”; 4) “NHBT-22”; 5) “NHBT-(N)19”; 6) “NHBT-template 22”. c) Separation of the products of covalent binding of holo-enzyme with “NHBT-16” (1), “NHBT-19” (2), and “NHBT-22” (3) after prolonged incubation in 8% SDS gels. d) Covalent binding of NHBT esters of oligonucleotides with core-enzyme after 1-h incubation: 1) “NHBT-(N)19”; 2) “NHBT-template 22”; 3) “NHBT-22”. The products were separated in 4% SDS gels. 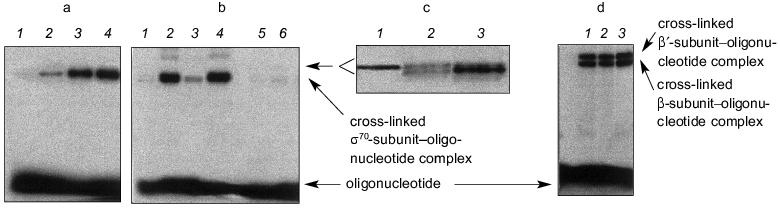
RNA polymerase is a multisubunit protein and therefore the NHBT ester of the oligonucleotide can covalently interact with one or several subunits, the character of interaction being dependent on whether the subunits belong to the holo- or core-enzyme. Under denaturing conditions of SDS-PAGE, individual subunits cross-linked with oligonucleotide are separated according to their mobility (mass). The yields of the cross-linked products were calculated based on the relationship between radioactivities of individual subunits containing covalently bound oligonucleotides and non-bound DNA.
Figure 2b shows the results of covalent binding of all the above described NHBT esters of oligonucleotides from the template and non-template strands in comparison with control oligonucleotide of statistical composition. As can be seen, both the NHBT ester of the statistical oligonucleotide (N)19 and “NHBT-template 22” do not covalently interact with holo-enzyme.
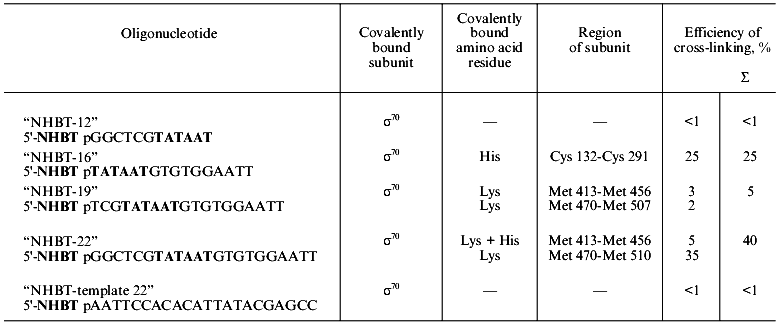
NHBT esters of oligonucleotides from the non-template strand of promoter (with the exception of “NHBT-12” devoid of a region after the TATAAT-block) covalently interact with holo-enzyme, displaying a different efficiency. Of the subunits of holo-enzyme, the sigma70-subunit participates predominantly in the formation of a covalent bond. “NHBT-12” is unable to form a covalent bond with the holo-enzyme. It should be noted that “NHBT-12” differs from other nucleotides from the non-template strand by the absence of the region that is adjacent to the TATAAT-block in the direction of initiation of transcription, and this is required evidently for sequence-specific interaction with the holo-enzyme. The results of covalent binding of NHBT esters of the studied oligonucleotides with holo-enzyme are summarized in the table.
Results of covalent binding of NHBT esters of oligonucleotides of various
composition with holo-enzyme of RNA polymerase
Covalent binding of all NHBT esters of oligonucleotides with core-enzymes is accompanied by the formation of bonds with the beta- and beta´-subunits of the protein (efficiency of binding is 10-12% for each subunit). Figure 2d shows the results of covalent binding of core-enzyme with “NHBT-(N)19”, “NHBT-template 22”, and “NHBT-22”. As is seen, covalent binding with core-enzyme is not sequence-specific.
The differences in covalent binding of NHBT esters of oligonucleotides with holo- and core-enzyme were demonstrated also with the data on competitive displacement of oligonucleotide “NHBT-22” from the complex with protein by nucleotides of different primary structure, namely by nucleotides of different length from the non-template strand of lac UV5 (“16” or “22”) and the template strand (complementary “22”) or (N)19-oligonucleotide of statistical composition. In the case of the holo-enzyme, addition of oligonucleotides from the non-template strand results in a significant decrease in the yield of the cross-linked product. The longer is the oligonucleotide, the greater is its effect. The decrease in the yield of the cross-linked product is less marked when oligonucleotide from the template strand was added. The oligonucleotide of mean-statistical composition has little or no effect on the yield of the product of cross-linking. This result provides support for the specificity of recognition in the complex with holo-enzyme. Cross-linking can be partially suppressed by addition of heparin (10 µg/ml) or double-strand 70-membered fragment of promoter T7A1 (in equimolar relationship with oligonucleotide).
In the case of covalent binding of “NHBT-22” with core-enzyme, addition of oligonucleotides of different composition also results in a decrease in the yield of the product of cross-linking. The effect of the competing oligonucleotide depends on its length, suggesting that binding is nonspecific. Complete suppression of binding occurs at concentration of heparin of 1 µg/ml, i.e., a concentration which is 10-fold less than it was with holo-enzyme.
Thus, covalent binding of NHBT esters of oligonucleotides with holo-enzyme is sequence-specific. The comparison of the results of covalent binding with the data on noncovalent binding indicates that the specificity of interaction of oligonucleotide with protein is independent of whether the oligonucleotide or its NHBT ester is used. This fact is a basis for using derivatives of such type in studying nucleic acid-protein interactions.
3. Identification of amino acid residues participating in covalent binding with a phosphate group of the oligonucleotide. Previously we showed that covalent binding of NHBT esters of oligonucleotides with proteins results in the formation of a covalent bond between a phosphate group of the oligonucleotide and nucleophilic groups of lysines or histidines of the protein [15]. Therefore, to locate a covalent bond between oligonucleotide and protein, it is necessary to identify the lysine or histidine of the protein participating in the formation of the covalent bond and specify the region of a subunit containing this amino acid residue. However, several lysines or histidines may be brought into the specified subunit region. Therefore, the next step in identification of the covalently bound amino acid residue could be point mutagenesis, i.e., replacing one of the proposed residues by a residue, which is unable of forming a covalent bond with activated phosphate group.
3.1. Resistance of the products of covalent binding of oligonucleotides with holo-enzyme to hydrolysis. Since a phosphoamide derivative with lysine and a phosphoimidazole derivative with histidine differ in chemical properties, the analysis of hydrolytic resistance of the product of cross-linking allows the amino acid residue entered into a reaction to be identified. The following conditions of hydrolysis were used: HCl solution, pH 2, where rapid cleavage of bonds of both types occurs, and MES buffer, pH 5, where 4-h incubation results in cleavage of only the bond between phosphate and histidine. Previously we proposed a simple method for discrimination between lysines and histidines. The method involves the treatment by 1-methyl imidazole at pH 8 [15]. This treatment results in rapid cleavage of the bond between phosphate and histidine (15-min incubation provides complete cleavage), whereas the bond between phosphate and lysine remains intact. Such conditions were used for treatment of cross-linked complexes.
5´-Activated phosphate of “NHBT-16” covalently interacts with histidine of the sigma70-subunit. This conclusion was confirmed by results of hydrolysis: the covalent bond was hydrolyzed both in HCl solution, pH 2, and in MES-buffer, pH 5.0. Besides, the product of cross-linking is rapidly cleaved as a result of transamination in 1-methyl imidazole (1-MeIm), pH 8.0 (Fig. 3). In cross-linked complex “19”-sigma70, lysine is essentially the sole residue involved in covalent binding. In cross-linked complex “22”-sigma70, a covalent bond is formed primarily with participation of lysine and to some extent with histidine. Results of identification of amino acid residues covalently bound with phosphate in all the complexes are summarized in the table.
3.2. Determination of the region of covalent binding of oligonucleotides with the sigma70-subunit of the holo-enzyme. We have specified the regions of the protein containing amino acid residues covalently bound with oligonucleotide. When separating the cross-linked complexes of holo-enzyme with “NHBT-16”, “NHBT-19”, and “NHBT-22” during SDS -PAGE in an 8% gel, we observed that rather long times of separation (2-2.5 h) provided splitting of the cross-linked sigma70 -“19” complex into two bands of comparable intensity. The sigma70-“22” complex is also separated into two bands, the lower of them comprising ~85% of the total intensity of the cross-linked complex. However, for the sigma70-“16” complex only one band was observed (Fig. 2c). We suppose that these bands correspond to the cross-linked complexes where phosphate group of the oligonucleotide is bound with different regions of the sigma70-subunit or different amino acid residues in the same region. To specify these regions and identify amino acid residues, we carried out preparative cross-linking and then analyzed the individual bands separated by SDS-PAGE.Fig. 3. Identification of the amino acid residue of the protein covalently bound with the 5´-phosphate of oligonucleotide “16” by hydrolysis of the cross-linked complex: 1) original cross-linked complex; 2-4) cleavage of complexes under the action of 0.01 M HCl; 5-7) cleavage of complexes under the action of 0.05 M MES, pH 5.0; 8-10) cleavage of complexes under the action of 0.05 M 1-methyl imidazole, pH 8.0. The separation was carried out by SDS-PAGE in an 8% gel. 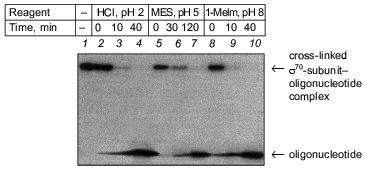
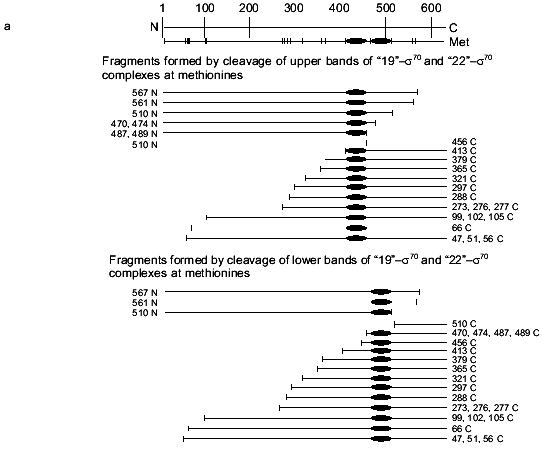
To specify the regions of subunits containing amino acid residues covalently bound with oligonucleotide, the cross-linked complex isolated from the gel after SDS-PAGE was cleaved by selective chemical reagents. Cleavage was conducted at methionines using BrCN or at cysteines using 2-nitro-5-thiocyanbenzoic acid under conditions where the protein molecule contains no more than one broken bond (as described in [16]). Such cleavage gives a set of shortened protein fragments, part of them being covalently bound with radioactively labeled oligonucleotide. In SDS-PAGE, these fragments are separated according to their masses (Figs. 4 and 5). To identify the fragments, we compared their mobilities in SDS-PAGE with the mobilities of markers of known masses. Comparison of fragments obtained by cleavage of the cross-linked complexes and fragments calculated theoretically for individual subunits allows the regions of attachment of oligonucleotide to a subunit to be specified with a precision within the limits of the distances between neighboring methionines and cysteines. Figures 4 and 5 show schematically the positions of methionines and cysteines in the sigma70-subunit and fragments formed during cleavage (Figs. 4a and 5a). Separation of fragments of the cross-linked complexes of the sigma70-subunit with “NHBT-16”, “NHBT-19”, and “NHBT-22” cleaved at methionines and cysteines is also shown (Figs. 4b and 5b).
Fig. 4. Determination of the region of the sigma70-subunit bound covalently with “16” by limited cleavage of the cross-linked complex at methionines and cysteines. a) Tentative scheme of cleavage of the sigma70-subunit into fragments at methionines and cysteines, respectively. The horizontal line of length 613 amino acid residues is a linear representation of the sigma70-subunit. The vertical lines on the horizontal line correspond to the positions of methionines and cysteines. The horizontal lines of different length given below are the N- and C-terminal fragments (N and C, respectively) formed by cleavage at corresponding methionines or lysines. The numbers near the lines refer to the number of the amino acid residue at which cleavage occurred. b) Separation of the products of cleavage of the sigma70-subunit cross-linked with “16” in 8% SDS-PAGE. Cleavage was conducted at methionines (2, 3) and cysteines (4, 5). The arrows point to the corresponding N-terminal fragments of the sigma70-subunit. The comparison of the bands in Figs. 4a and 4b allowed the region of the sigma70-subunit bound covalently with 32P-labeled “16” to be specified. Circles in panel (a) refer to histidines, one of which is involved in the formation of a covalent bond.
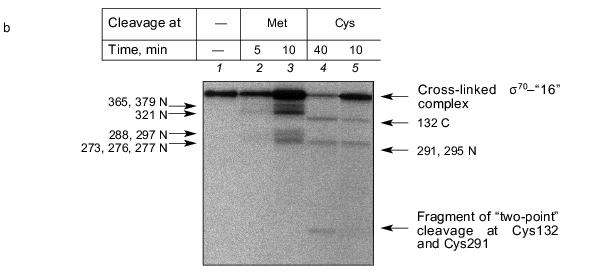
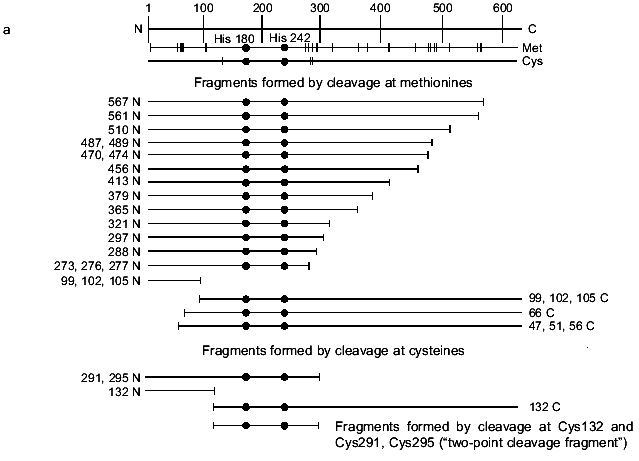
The results show that the 5´-phosphate group of the oligonucleotide in the cross-linked “16”-sigma70 complex is linked with histidine in the region between Cys132 and Cys291. In this region only two histidines are located: His180 and His242. In the “19”-sigma70 complex a covalent bond is formed with the participation of one of the lysines located between Met413 and Met456 in the case of the upper band in Fig. 2c, which comprises 60% of the total amount of bound protein, and between Met470 and Met507 in the case of the lower band in Fig. 2c. In the “22”-sigma70 complex, a covalent bond is formed with the participation of a lysine located between Met470 and Met507 in the case of the major lower band in Fig. 2c (85% of the total amount of bound protein) and with the participation of lysine or histidine located between Met413 and Met456 in the case of the minor upper band. These regions contain several lysines (414, 418, 426, 493, 496, 499, 502) and His455.Fig. 5. Determination of the region of the sigma70-subunit bound covalently with “19” and “22” by limited cleavage of the cross-linked complex at methionines. a) Tentative scheme of cleavage of the sigma70-subunit into fragments at methionine. The horizontal line of length 613 amino acid residues is a linear representation of the sigma70-subunit. The vertical lines on the horizontal line correspond to the positions of methionines. The horizontal lines of different length given below are the N- and C-terminal fragments (N and C, respectively) formed by cleavage at the corresponding methionines. Numbers near the lines refer to the number of the amino acid residue at which cleavage occurred. b) Separation of the products of cleavage of the sigma70-subunit bound covalently with “19” and “22” in gradient (8-16%) SDS-PAGE. The cross-linked sigma70-“19” complex: upper band of lane 2 in Fig. 2c (1-3) and lower band of lane 2 in Fig. 2c (4-6). The cross-linked sigma70-“22” complex: upper band of lane 3 in Fig. 2c (7-9) and lower band of lane 3 in Fig. 2c (10-12). The arrows point to the corresponding N- and C-terminal fragments of the sigma70-subunit. The comparison of the bands in Figs. 5a and 5b allowed the regions of the sigma70-subunit bound covalently with 32P-labeled “19” and “22” to be specified. Ellipses in panel (a) refer to the regions of the sigma70-subunit bound covalently with “19” and “22”.
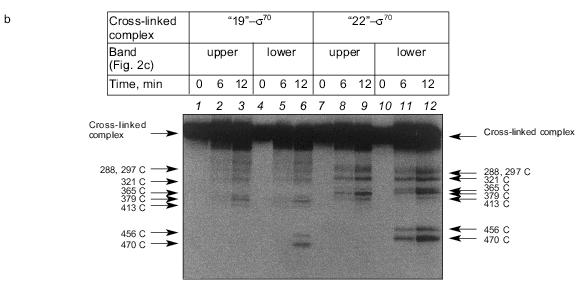
Based on the results of cross-linking, we propose a scheme for the mutual arrangement of the non-template strand of the promoter and the sigma70-subunit possessed by holo-enzyme in the open complex (Fig. 6). X-Ray study of the complex is yet to be carried out. Therefore, when drawing the scheme we used the spatial structure of a fragment of the sigma70-subunit determined in [20] by the X-ray method (residues 114-446), the data on the contacts of protein with DNA [4, 6, 21, 22], and our data on the contacts of phosphate groups.
The above fragment of the sigma70-subunit contains lysines and histidines, which, according to our data, covalently interact with activated phosphate groups at positions -12, -15, and -18 of the oligonucleotides. The amino acid residues coming in contact with phosphate -15 and -18, namely lysines located between Met470 and Met507 and His455, are situated outside the region of the sigma70-subunit with known spatial structure.Fig. 6. Scheme illustrating the mutual arrangement of oligonucleotide and sigma70-subunit possessed by the holo-enzyme. The structure of the sigma70-subunit [20] is designated by the black color. alpha-Helices 13 and 14 are indicated by zigzag. The white line is the oligonucleotide with marked off positions -12, -15, and -18. Contacts of the 5´-phosphate groups with lysines and histidines of the sigma70-subunit determined in the present work are denoted by diffuse white arrows. Contacts of heterocyclic bases with amino acid residues of the sigma70-subunit [4, 6, 21, 22] are designated by thin arrows. 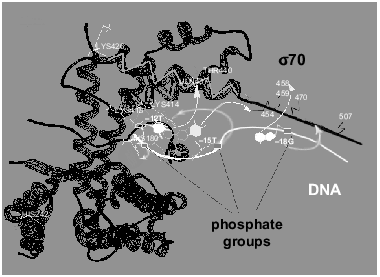
It is known that helices 13 (residues 401-418) and 14 (residues 427-445) of the sigma70-subunit are responsible for specific binding of DNA [4, 6, 20]. Taking into account this fact and the data on the contacts of heterocyclic bases [4, 6, 21, 22], we arranged the oligonucleotide strand along these helices. The results are in good agreement with the contacts of phosphate groups determined by us.
The 5´-phosphate group at positions -15 and -18 (oligonucleotides “19” and “22”) interacts with one of the lysines (414, 418, or 426) located in the region 413-456 possessed by one of the helices (13 or 14).
Of two histidines located between Cys132 and Cys291 and bound covalently at position -12 of oligonucleotide (“16”), His180 is located significantly closer to helices 13 and 14 than His242 (17-27 A and 42-47 A, respectively). The lesser distance corresponds to the distance between phosphate groups at positions -12 and -15 separated by three nucleotides in the oligonucleotide. Therefore, we suppose that it is precisely His180 that interacts with phosphate at position -12.
As pointed out above, for lack of data of X-ray studies for the corresponding region of the sigma70-subunit, localization of the contact of phosphate group -18 (oligonucleotide “22”) is impossible. However, effective cross-linking (40%) with lysine from the region between Met470 and Met507 indicates that the phosphate group and this lysine are brought into proximity.
4. Mapping the regions of covalent binding of NHBT-activated nucleotides with core-enzyme. When mapping the product of covalent binding of “NHBT-16” with core-enzyme, we showed that covalent binding with the beta- and beta´-subunits proceeds with the same efficiency (10%). For the beta-subunit, lysine or histidine in the region between Met1232 and Met1243 undergoes phosphorylation. For the beta´-subunit, phosphorylation of a residue between Cys71 and Cys88 occurs. “NHBT-22” interacts covalently with the same regions of the beta- and beta´-subunits with efficiency of 12% in each case.
The region between Met1232 and Met1243 of the beta-subunit contains the conservative residue His1237, which is involved in the formation of the active site of the enzyme [23] and, as shown earlier, interacts with alpha-phosphate of the initiating substrate [24]. This region of the beta-subunit may covalently interact with photoactivated analogs of DNA during elongation [25]. Our data suggest that the beta-subunit participates in nonspecific interaction with single-strand DNA.
The region of the beta´-subunit forming a covalent bond with the oligonucleotide is a part of a “zinc finger”. This structural element is typical for DNA-binding proteins. Studies of elongation [25] showed that under certain conditions a fragment of double-strand DNA is capable of interacting covalently with the beta´-subunit in the region between Met30 and Met102. Thus, our data provide support for the DNA-binding function of the region of the beta´-subunit under discussion.
The results suggest that NHBT esters of oligonucleotides, which are used for the first time in studying the model of the open promoter complex, help to elucidate the contacts of phosphate groups of oligonucleotide with nucleophilic groups of lysines and histidines of the protein.
The use of these derivatives allows the efficiency of cross-linking in the complex to be enhanced to 40%. The specificity of binding with protein is unaffected by substitution of NHBT ester of oligonucleotide for oligonucleotide. Covalent binding of these oligonucleotide derivatives proceeds regioselectively, namely, with lysines and histidines immediately adjacent to activated phosphate. In contrast to the majority of previously used derivatives, in this case it is the phosphate group of an oligonucleotide that directly participates in the formation of a covalent bond. Owing to the absence of a spacer group in the conjugate, the contacts of phosphate groups may be localized with high precision. Our data give a more accurate view of the structure of the open complex and are of great importance since X-ray studies of the structure of the complex of DNA with the sigma70-subunit possessed by the holo-enzyme are not yet available.
This study was funded by grants 99-03-33127a, 00-04-48260, and 00-15-97944 from the Russian Foundation for Basic Research.
REFERENCES
1.Helmann, J. D., and Chamberlin, M. J. (1988)
Annu. Rev. Biochem., 57, 839-872.
2.Hawley, D. K., and McClure, W. R. (1983) Nucleic
Acids Res., 11, 2237-2255.
3.DeHaseth, P. L., and Helmann, J. D. (1995) Mol.
Microbiol., 16, 817-824.
4.Owens, J. T., Chmura, A. J., Murakami, K., Fujita,
N., Ishihama, A., and Meares, C. F. (1998) Biochemistry,
37, 7670-7675.
5.Roberts, C. W., and Roberts, J. W.(1996)
Cell, 86, 495-501.
6.Bown, J. A., Owens, J. T., Meares, C. F., Fujita,
N., Ishihama, A., Busby, S. J., and Minchin, S. D. (1999) J. Biol.
Chem., 274, 2263-2270.
7.Savinkova, L. K., Baranova, L. V., Knorre, V. L.,
and Salganik, R. I. (1988) Mol. Biol. (Moscow), 22,
807-812.
8.Marr, M. T., and Roberts, J. W. (1997)
Science, 276, 1258-1260.
9.Tulokhonov, I. I., Savinkova, L. K., Rau, V. A.,
and Rar, V. A. (1999) Mol. Biol. (Moscow), 33,
169-173.
10.Fedoriw, A. M., Liu, H., Anderson, V. E., and
deHaseth, P. L. (1998) Biochemistry, 37, 11971-11979.
11.Heyduk, E., and Heyduk, T. (1999) J. Biol.
Chem., 274, 3315-3322.
12.Kulbachinskiy, A., Mustaev, A., Goldfarb, A., and
Nikiforov, V. (1999) FEBS Lett., 454, 71-74.
13.Chenchik, A. A., Bibilashvili, R. Sh., and
Mirzabekov, A. D. (1981) Doklady RAN, 260, 765-768.
14.Chenchik, A. A., Bibilashvili, R. Sh.,
Mirzabekov, A. D., and Shik, V. V. (1982) Mol. Biol. (Moscow),
16, 35-46.
15.Taran, E. A., Ivanovskaya, M. G., Gait, M. J.,
and Shabarova, Z. A. (1998) Mol. Biol. (Moscow), 32,
832-839.
16.Markovtsov, V., Mustaev, A., and Goldfarb, A.
(1996) Proc. Natl. Acad. Sci. USA, 93, 3221-3226.
17.Bergess, R. R., and Jendrisak, J. J. (1975)
Biochemistry, 14, 4634-4638.
18.Borukhov, S., and Goldfarb, A. (1993) Protein
Express. Purif., 4, 503-511.
19.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, 2nd ed., Cold
Spring Harbor Laboratory Press, New York.
20.Malhotra, A., Severinova, E., and Darst, S. A.
(1996) Cell, 87, 127-136.
21.Siegele, D. A., Hu, J. C., Walter, W. A., and
Gross, C. A. (1989) J. Mol. Biol., 206, 591-603.
22.Waldburger, C., Gardella, T., Wong, R., and
Susskind, M. M. (1990) J. Mol. Biol., 215, 267-276.
23.Mustaev, A., Kashlev, M., Lee, J., Polyakov, A.,
Lebedev, A., Zalenskaya, K., Grachev, M., Goldfarb, A., and Nikiforov,
V. (1991) J. Biol. Chem., 266, 23927-23931.
24.Grachev, M. A., Lukhtanov, E. A., and Mustaev, A.
A. (1990) Bioorg. Khim., 16, 1379-1385.
25.Nudler, E., Avetissova, E., Markovtsov, V., and
Goldfarb, A. (1996) Science, 273, 211-217.