REVIEW: Active Site Interactions in Oligomeric Structures of Inorganic Pyrophosphatases
S. M. Avaeva
Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3181; E-mail: avaeva@genebee.msu.su
Received October 11, 1999
Recent progress in studies of the mode of action of cytoplasmic inorganic pyrophosphatases is mainly due to the analysis of a dozen and a half structures of the apoenzyme, its complexes, and mutants. However, despite considerable research on the mechanism of action of these enzymes, many important problems remain unclear. Among them is the problem of active site interactions in oligomeric structures and their role in catalysis; this review focuses on this problem. The abundant experimental data requires generalization and comprehensive analysis. A characteristic feature of the spatial structure of inorganic pyrophosphatases is a flexible system of noncovalent interactions between protein groups penetrating the whole molecule of the oligomeric enzyme. Binding of metal ions, sulfate (an analog of the product of the enzymatic reaction), and affinity phosphorus-containing inhibitors at the active site or site-directed mutagenesis induce rearrangements in the set of hydrogen and ionic interactions, which change active site properties and in some instances, cause molecule asymmetry. In the trimeric form of Escherichia coli pyrophosphatase obtained by dissociation of a hexamer, active sites also interact with each other, which is manifested by negative cooperativity upon substrate binding. The association of trimers into the hexamer leads to perfect organization of active sites and to their coordinated functioning, probably due to the restoration of communication channels between the trimers.
KEY WORDS: inorganic pyrophosphatase, active site interactions, trimeric enzyme form, X-ray analysis, site-directed mutagenesis
In the last five years, considerable advances have been made in studies of the mode of action of cytoplasmic inorganic pyrophosphatases (PPases). Since these enzymes are closely related to the synthesis of vital biopolymers, they can be isolated from all organisms. The primary structures of more than three dozen PPases have been determined, and this number rapidly increases with successful sequencing of a large number of genomes. Sequence homology varies considerably; however, a dozen and a half amino acid residues exposed to the active site cavity are conserved. In 1998 soluble PPases whose amino acid sequences showed no sequence homology with known enzymes were discovered [1]. They exhibit all specific properties of PPases and probably originated by convergent evolution. All PPases function as homooligomers. Procaryotic enzymes are hexamers with subunit molecular mass of .20 kD. Eucaryotic PPases are dimers with subunits of .30 kD.
Determination of PPase three-dimensional structure was an important stage in PPase analysis necessary to gain a more penetrating insight into their functions. The first 3-D structure was solved at the Institute of Crystallography (Russian Academy of Sciences, Moscow) in 1981 for Saccharomyces cerevisiae PPase (Y-PPase) [2]. This work formed the basis for further progress in determination of PPase 3-D structures. Three more 3-D structures of PPases from Escherichia coli (E-PPase) [3-5], Thermus thermophilus [6], and Sulfolobus acidocaldarius [7] have been reported. Despite different size of subunits in procaryotic PPases, their spatial organization is very similar, and excess amino acid sequences of the heavier subunits form polypeptide loops on the surface of the molecule.
In 1995-1999, X-ray analysis of E-PPase complexed with manganese ion [8], one [9] and two magnesium ions [10, 11], and with sulfate, the phosphate analog [12], at the active site was performed. Y-PPase complexed with four manganese ions and two phosphates was described [13, 14]. The three-dimensional structures of six mutant PPases were solved [15, 16]. Some of these structures will be discussed in more detail in this review.
PPi, a simple and very reactive compound, is a substrate of inorganic pyrophosphatase. Therefore, a priori its catalytic hydrolysis seemed rather simple, but this is not the case, as we shall show later. It should be noted that bivalent metal ions are necessary for catalysis. Magnesium is the most effective: this effect decreases as follows: Mn2+ > Zn2+ > Co2+ > Cd2+. The hydrolysis rate of other substrates is much lower, and the catalytic reaction requires different conditions. Thus, tripolyphosphate is hydrolyzed by Y-PPase in the presence of Mg2+ at alkaline pH [17]; hydrolysis of pyrophosphoric acid esters and ATP proceeds in the presence of Zn2+ or Mn2+ [18, 19].
The maximum hydrolysis rate is observed when in addition to PPi, 3-4 metal ions (M) bind to the active site. Two of them associate with the enzyme before PPi binding and occupy sites I and II with binding constants differing tenfold. Site III accepts M as a result of MgPPi binding. One more site, site IV, is filled with the M simultaneously with the products of substrate hydrolysis bound at the active site.
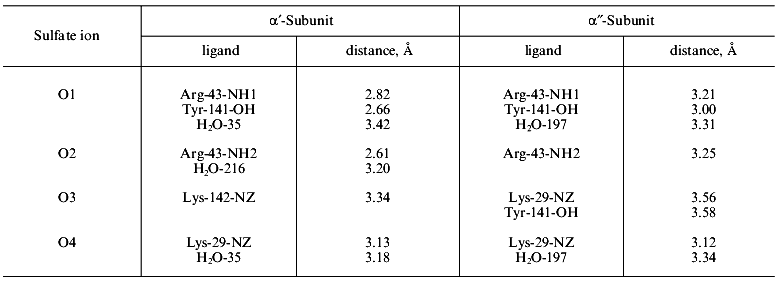
In the active site cavity, eight dicarboxylic amino acid residues (Asp-42 (71), Asp-65 (115), Asp-67 (117), Asp-70 (120), Asp-97 (147), Asp-102 (152), Glu-20 (48), and Glu-31 (58)), arginine (Arg-43 (78)), three lysine (Lys-29 (56), Lys-104 (154), and Lys-142 (193)), and two tyrosine residues (Tyr-55 (93) and Tyr-141 (192)) are located. Their positions correspond to the E-PPase sequence; the positions of amino acid residues in the Y-PPase sequence are given in parentheses. All these amino acid residues are involved in binding of metal ions and/or substrate.
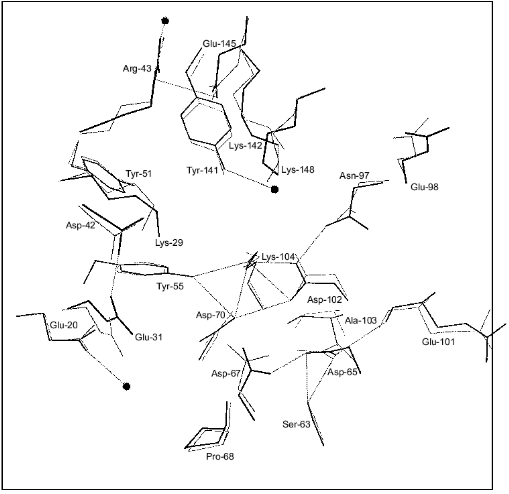
Figure 1 shows schematically the location of four metal ions and two phosphates in the Y-PPase active site. At M(I), Asp-152, Asp-115, and Asp-120 are protein ligands of Mn2+. The last residue also binds Mn2+ at M(II) as its single protein ligand. Upon substrate binding, both metal ions are coordinated with pyrophosphate oxygen atoms. M(III) is a “substrate” metal coordinated with two phosphates from pyrophosphate and with two protein groups, Asp-147 and Asp-152. M(IV) is bound to the oxygen atoms of two phosphates and one protein ligand, Glu-58. The phosphate groups are fixed both by metal ions and several protein functional groups.
The arrangement of metal ions in the active site may indicate their multiple functions. Metal ions provide the necessary conformation for substrate binding and determine the degree of binding. The contacts of phosphate groups with the metal, in turn, increase electrophilicity of the phosphorus atom that is essential for efficient hydrolysis. The key role of metal ions consists in generating a nucleophile by inclusion of water into their coordination sphere, thus decreasing its pKa and in stabilizing the transition state.Fig. 1. Disposition of phosphates and metal ions at the active site of Y-PPase. Manganese ions, whose coordination spheres are filled with water molecules to n = 6, are shown in squares. Water molecules are designated by small circles; only those which interact both with M and other active site functional groups are shown. The contacts of metal ions are shown with solid lines, and hydrogen bonds are designated by dotted lines.
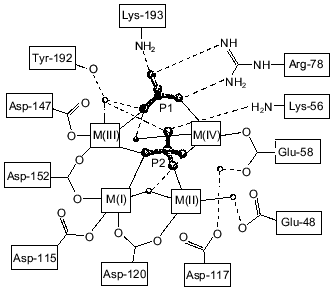
Several hypotheses on the mechanism of substrate hydrolysis have been advanced [13, 14]. They are based either on attempts to identify the phosphate oxygen introduced with the associating water molecule in the course of hydrolysis or to detect a water molecule that has already occupied the necessary position for a subsequent hydrolysis act before the hydrolysis products leave the active site. The weak point in these hypotheses is the assumption that at the active site the binding sites of phosphorus from phosphate, the reaction product, and the attacked phosphorus from pyrophosphate coincide. Recent findings indicate that this is not the case [20].
Despite considerable progress in the understanding of PPase action, a number of important problems remain obscure. The conformation of pyrophosphate at the active site, the structure of the substrate transition state during its conversion to reaction products, as well as enzyme structural rearrangements in the course of catalysis are unknown. The order of phosphate release from the active site is unclear; the available data are conflicting. It is still incompletely understood how a nucleophilic particle necessary for catalysis is generated and which factors determine the variation in properties of PPases isolated from different sources.
In addition to problems directly connected with active site functioning, another important problem of active site interactions in oligomeric PPase structures and a closely related problem of activity regulation mechanisms deserve special attention. These problems still remain beyond the scope of research of most scientists concerned with PPases. This review is the first attempt to summarize our and literature data on this subject and thus to draw attention to the problem.
E-PPase STRUCTURE AND INTRAMOLECULAR INTERACTIONS
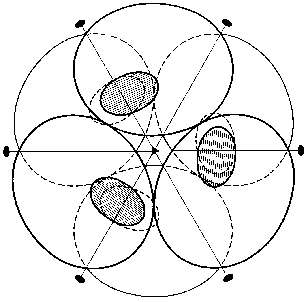
The spatial structure of PPases provides the basis for a permanent set of interactions involving the entire oligomeric molecule. We shall mainly focus on the E. coli enzyme. The hexameric molecule of E-PPase is composed of two trimers [3]. The two trimers are located in parallel planes turned relative to each other (Fig. 2). Each subunit forms contacts with four adjacent subunits. In the trimer the contacts are mainly stabilized by hydrophobic interactions and hydrogen bonds, with peptide bonds of the protein backbone acting as donors and acceptors. Antiparallel symmetric alpha-helical regions largely contribute to intertrimeric contacts. The interactions between them are stabilized by two ionic pairs, His-136-Asp-143´ and His-136´-Asp-143, and a hydrophobic contact, His-140-His-140´. Other intertrimeric contacts include the regions Ser-46-Phe-50 and Asn-24-Asp-26. The hexamer dissociation to trimers occurs at pH below 6 [21]. The interactions between subunits in the trimer are stronger than intertrimeric contacts, and in all likelihood the monomer cannot be obtained without special modification of groups involved in intertrimeric contacts.
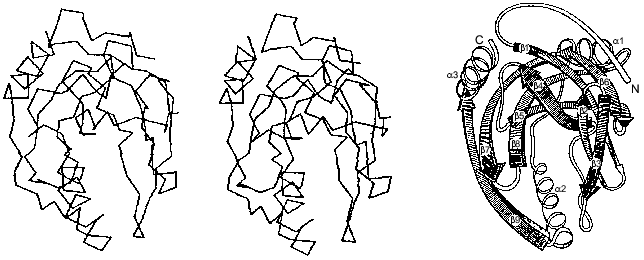
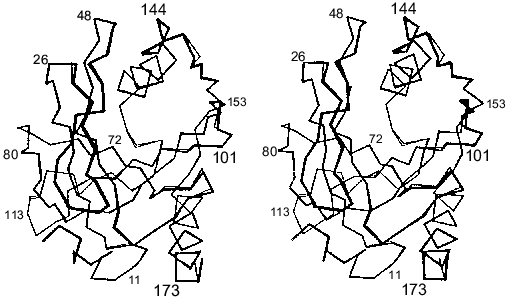
The polypeptide backbone consisting of 175 amino acid residues is shown in Fig. 3. Two extended alpha-helices and nine beta-strands, some of which form a barrel structure, are characteristic of the enzyme secondary structure. A large internal cavity in Fig. 3 corresponds to the enzyme active site. One side of the active site is formed by alpha-helix, whose opposite side is involved in intertrimeric contacts.Fig. 2. Quaternary structure of hexameric E-PPase. Three crystallographic second-order axes (dark ellipses) and a third-order axis (dark triangle) are shown. Hatched regions indicate the position of the active site in the upper trimer, and symmetric dotted regions, in the lower trimer.
A whole complement of noncovalent interactions between amino acid residues within subunits and between them in a hexamer plays a major role in stabilizing the native conformation of the molecule [12]. Peptide groups are linked by approximately 80 hydrogen bonds. In addition, 86 polar functional amino acids can act as hydrogen bond donors and/or acceptors and form ion pairs. Actually, several dozen interactions are realized in each subunit. It is evident that 150 water molecules found in the enzyme subunit also participate in a branched system of hydrogen bonds.Fig. 3. Packing of the polypeptide chain in the E-PPase subunit (spatial and schematic representations).
A specific feature of E-PPase organization is the existence of flexible regions that are less structured than the remaining molecule and are loosely bound to the stable core. This primarily refers to the regions Ser-63-Asp-67, Lys-94-Asp-102, and Glu-145-Glu-153. These sequences include the following active site groups: Asp-65, Asp-67, Asp-97, and Asp-102. Such principle of active site organization and its evolutionary stability suggest that flexibility of these regions is essential for enzymatic activity. It may provide the active conformation for substrate binding and release of the products of the enzymatic reaction. Note that all mobile fragments of the polypeptide chain contain glycine residues: Gly-66, Gly-100, and Gly-147.
Since ligand-binding sites at the active site are located just between the flexible regions, their filling may lead to loop movement and to further rearrangements in noncovalent interactions due to disruption of some bonds and formation of others.
STRUCTURAL CHANGES INDUCED BY POINT MUTATIONS OF THE ACTIVE SITE
GROUPS
Genetic engineering as a tool for site-directed mutagenesis was supposed to provide a promising and accurate method for identifying functions of particular amino acids. This method replaced chemical modifications, whose weak points were evident. Even a highly specific chemical reagent used for modification is an additional component introduced into the enzyme molecule and therefore may play a pivotal role in changing properties of the protein. However, analysis of mutant PPases clearly showed that changes in catalytic properties are not due to single substitutions of amino acids, but represent a complex response of the molecule induced by the rearrangement of noncovalent interactions between numerous protein groups.
About 40 different mutant forms of E-PPase and Y-PPase with single substitutions of active site amino acid residues have been obtained [22-24]. Dicarboxylic amino acids were substituted either for corresponding amides (without changing the size of the residue) or for the homologous dicarboxylic acids (with preservation of charge); lysine was substituted for arginine and vice versa; tyrosine was replaced by phenylalanine. In 1998, the three-dimensional structures of four mutant E. coli proteins (with substitutions Asp-42-Asn, Asp-65-Asn, Asp-70-Asn, and Asp-97-Asn) [15] and two mutant Y-PPases (Arg-78-Lys and Asp-117-Glu) [16] were solved.
A detailed study of the mutant enzyme properties showed that all substitutions (except for Asp-42-Asn E-PPase [25]) decrease the catalytic constant of the hydrolysis rate and change the pKa of enzyme groups involved in catalysis, but only slightly affect affinity for the substrate and metal. This observation was quite unexpected. Actually, the role of particular residues is quite different. For instance, Lys-29 (56) forms a contact with phosphate oxygen (Fig. 1), which in turn, is fixed in the active site by a set of other interactions. However, the Lys-29 (56) -->_Arg substitution in E. coli and yeast enzymes leads to a decrease in catalytic activity to 7% and 9% of WT PPase activity, respectively, at saturating substrate concentration [26]. One more example--substitution of a single magnesium protein ligand at the second E-PPase site (Asp-70-Asn) decreases Mg2+ affinity for the enzyme only twofold; however, the rate catalytic constant decreases more than a hundred times [23].
The consequences of single amino acid substitutions at the active site become clear from the comparison of Asp-97-Asn and Asp-97-Glu substitutions in E-PPases and Asp-147-Glu replacement in Y-PPase. The properties of these mutant forms are well studied: the dependences of kcat on pH and dissociation constants of protein groups affecting catalytic efficiency were measured [23, 26]. For Asp-97-Glu E-PPase, the kinetic constants of all steps of the catalytic reaction (substrate binding, conversion, and release of the reaction products) were estimated [27]. Finally, the three-dimensional structure of Asp-97-Asn E-PPase at 2.2 Å resolution was solved [15]. Conserved in all PPases, this amino acid residue participates in binding Mg2+, which enters the active site with the substrate--M(III) (Fig. 1). The second protein ligand for the metal ion is Asp-102 (152). Therefore, one could expect that Asp-97 (147) substitution would interfere with substrate binding. However, as it follows from Table 1, any substitution changes Km not more than ninefold, but the rate constant varies considerably and decreases two hundred times. Analysis of kinetic and thermodynamic parameters of PPi hydrolysis catalyzed by Asp-97-Glu E-PPase demonstrated that the mutation affects all stages of the catalytic reaction: the rates of PPi hydrolysis and synthesis decrease, and the rate of release of hydrolysis products (of both phosphates) from the active site also decreases. To explain these results, the cooperation of the active site groups and their perturbation caused by the amino acid substitution was postulated [27]. It also seemed tempting to explain changed properties of mutant proteins by a decrease in pKa of the major enzyme group responsible for the efficiency of hydrolysis [22]. However, Table 1 shows that there is no correlation between a decrease in enzyme activity and changes in pKEH2 and pKESH2, compared to WT PPase.
Table 1. Kinetic parameters of mutant PPases
with an Asp-97 (147) substitution
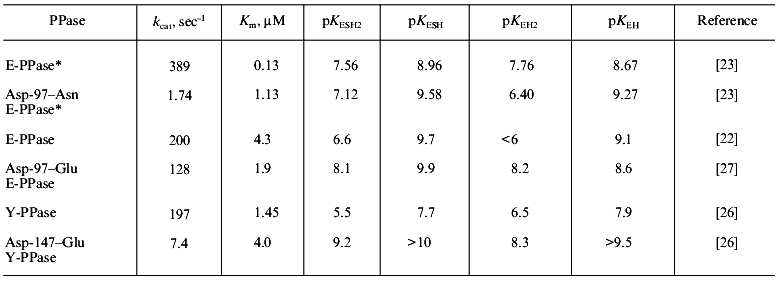
*For this enzyme, all parameters were measured at Mg2+ concentration of 5 mM; in other instances, Mg2+ concentration was 20 mM.
The fallacy of the scenario postulating only local changes in the active site groups induced by mutations was shown by X-ray analysis [15]. The comparison of polypeptide backbones of the mutant and native proteins reveals their identity except for the region 146-153; the displacement of this region leads to the formation of new hydrogen bonds between peptide groups (Fig. 4). The functional groups are shifted much more, and about 20 of them are displaced by more than 1.5 Å. Figure 5 shows the superposition of the mutant E-PPase active site structure on the apoenzyme. Most groups are shifted; however, it is important that Asn-97 also changed position, although it retained virtually all contacts. The Asp-97 carboxyl group interacted with Asp-102 carboxyl (the spacing 2.8 Å) and with nitrogen atoms from peptide groups of Ala-99 (2.6 Å) and Glu-98 (2.6 Å). The amide group of Asn-97 also preserved these contacts (3.0, 2.9, and 3.0 Å, respectively).
Fig. 4. Comparison of the polypeptide chain packing in Asp-97-Asn E-PPase (black color) subunit and apoenzyme (gray color). The numbers indicate positions of amino acid residues in the polypeptide chain.
One more spectacular manifestation of distant consequences of mutations in the active site consists in weakening of intertrimeric contacts, and hence, in an increased dissociation constant of the hexameric enzyme [28]. The opposite situation deserves special attention. Amino acid substitutions in the contact zone of trimers (His-136-Gln and His-140-Gln E-PPases) increase the rate constants to 225 and 110%, respectively, as compared to WT PPase [29]. This example shows that without structural data, site-directed mutagenesis may be misleading in determining the role of mutated residues.Fig. 5. Superposition of the active site groups in Asp-97-Asn E-PPase (black color) and apoenzyme (gray color). The molecules of the solvent are designated by circles; the functional groups of the mutant PPase located at a distance not exceeding 3.3 Å are connected with dotted lines.
Thus, investigations of mutant PPases provided abundant data in support of the notion that the native conformation of enzymes is determined and maintained by a whole complement of noncovalent interactions penetrating the entire molecule.
STRUCTURAL CHANGES INDUCED BY LIGAND BINDING TO THE PPase ACTIVE
SITE
The concept postulating flexibility of PPase structure and its response to mutations of the active site functional groups is confirmed by the introduction of different ligands to the PPase active site. Metal ions, sulfate (the phosphate analog, which is the product of enzymatic reaction) and affinity inhibitors (phosphoric acid esters) will be considered as ligands.
PPase interaction with metal ions. Early studies of enzyme saturation with metal ions demonstrated that binding is accompanied by conformational changes shown by the appearance of difference UV and fluorescence spectra; this formed the basis for estimating metal affinity for the enzyme [30]. Later it became evident that structural rearrangements are not confined to the active site, but involve other regions of the molecule. The effect of Mg2+ on intertrimeric contacts, when both the dissociation constant of the hexamer and association constant of trimers change, illustrates this phenomenon [28, 29].
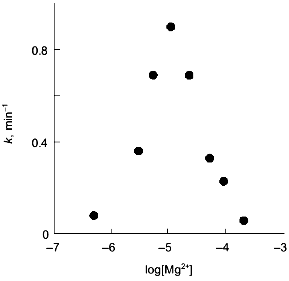
The observations demonstrating the interactions between metal-binding sites are of particular interest. Thus, the influence of Ca2+-binding sites on each other becomes evident from equilibrium dialysis [20]. Since the cooperativity of Ca2+ binding is preserved during the dissociation of a hexamer to trimers, interactions between subunits in the trimer were suggested. The interactions between Mg2+-binding sites are observed during Y-PPase inhibition with hydroxylamine in the presence of increasing Mg2+ concentrations [31]. The relationship indicates the presence of two Mg2+-binding sites with similar affinity for magnesium in the enzyme molecule (Fig. 6). However, the filling of these sites produces opposite effects: binding to the first site increases the rate of inactivation, while the saturation of the second site eliminates this effect.
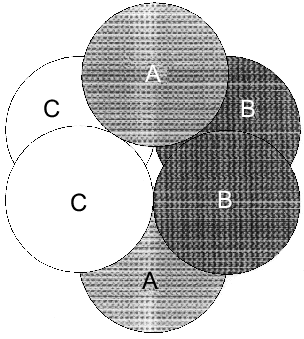
However, as shown in the previous section, the most convincing evidence for the induction of subunit asymmetry during the filling of metal-binding sites is based on X-ray data [10, 11]. The crystals of E-PPase complexed with magnesium ions were grown in the presence of ammonium chloride. Ammonium ions compete with Mg2+; therefore, Mg2+ concentration used for crystallization was rather high and exceeded the dissociation constant for the low-affinity site by two orders of magnitude. However, even this concentration was not sufficient to saturate all binding sites in the hexamer. This favored the detection of the transition state of the complex. The main characteristic feature of the complex was that it contained three types of subunits--A, B, and C. These subunits differed both in structure and the degree of saturation of the second site with magnesium. In subunit A, this site was completely saturated; in subunit B, only by 70%, and in subunit C, it was less than 20% saturated. This fact definitely indicates the existence of cooperative interactions between subunits. Alternatively, if Mg2+ concentrations were not sufficient for saturating all sites, they would not be filled completely, but the degree of saturation would be similar for all subunits. The presence of three types of subunits of different structure changes the symmetry group of the crystal with loss of third- and second-order axes (Fig. 7).Fig. 6. Effect of Mg2+ on the rate constant of Y-PPase inhibition with hydroxylamine. Mg2+ concentration is given in moles per liter.
The data presented suggest that metal binding to the active site induces subunit asymmetry due to interactions between PPase active sites.Fig. 7. Scheme of hexameric packing of nonequivalent subunits of E-PPase complexed with Mg2+.
Structure of E-PPase complexed with sulfate. The sulfate complex was obtained by crystallization of E-PPase in the presence of ammonium sulfate. X-Ray analysis at 2.2 Å resolution showed that each subunit contains one sulfate at the active site [12]. The comparison of the sulfate complex with Y-PPase complexed with Mn2+ and Pi [13] confirmed that the site of sulfate binding coincides with that of one phosphate. Similar to phosphate, sulfate is surrounded by basic amino acid residues (Arg-43, Lys-142, Lys-29, and Tyr-141). Two most important features characterize the sulfate complex. First, sulfate binding initiates significant reorganization of the entire complement of intramolecular interactions in each subunit, and second, these structural changes are different in the two trimers of a hexamer. As a result, six chemically identical subunits of the apoenzyme (alpha6) are transformed to the structure: alpha´3alpha´´3. Structural alterations in subunits within the complex affect the packing of molecules in the crystal and change unit parameters.
The comparison of sulfate complex and apoenzyme structures (Fig. 8) shows that 16 groups of the active site changed their position to a greater or lesser extent. The NH1- and NH2-groups of Arg-43 shifted most (6-7 Å). In the free enzyme, the guanidium group of arginine was exposed to the solvent, but in the presence of sulfate it is found at the active site. In two types of subunits, alpha´ and alpha´´, sulfate is associated differently with protein functional groups. This is shown in Table 2. The bonds between sulfate and Arg-43 in the alpha´-subunit are shorter than in the alpha´´-subunit. In the alpha´-subunit there is a contact with Lys-142, while this bond is absent from the alpha´´-subunit. The bond with Tyr-141 is shorter in the alpha´´-subunit than in the alpha´-subunit. The conversion of apoenzyme to the alpha´3alpha´´3 structure is accompanied by rearrangements of internal interactions extending far beyond the active site. Thus, nine hydrogen bonds between peptide groups present in the apoenzyme are destroyed, while 20 and 19 new bonds appear in the alpha´- and alpha´´-subunits, respectively. More significant changes occur in protein functional groups. While 17 and 14 contacts disappear, 30 and 35 new bonds are formed in the alpha´- and alpha´´-subunits, respectively.
Table 2. Protein ligands of sulfate in the
alpha´- and alpha´´-subunits of
E-PPase
It should be noted that the order of events could be opposite: changes in contacts in the crystal and, consequently, modification of the molecule structure. Such suggestion is inconsistent with observations made in [5]. Crystallization of E-PPase in the presence of small amounts of substrate analog or without it (other crystallization conditions were held constant) led to different packing of molecules in the crystal unit. Although the concentration of the analog was insufficient for saturating the active sites and complex formation, during long crystallization process, due to constant association and dissociation, the analog could change conformation of all protein molecules. These experiments show that conformational transitions occur slowly.Fig. 8. Superposition of the active site groups in the alpha´´-subunit of E-PPase complex with sulfate (black color) and apoenzyme (gray color). The solvent molecules are designated by circles.
Thus, binding of sulfate (phosphate analog) causes conformational changes in subunits involving the whole molecule and leading to asymmetry of trimers in the hexamer.
Reaction of PPase with phosphoric acid esters. Only a limited number of agents capable of changing PPase activity that can be assigned to natural regulators of the enzyme are presently known. These are primarily Ca2+: at low concentrations, Ca2+ binding at the M(II) site leads to enzyme inhibition [20]; and also phosphoric acid monoesters, a group of compounds involved in cellular metabolism.
Methyl phosphate, phosphoglycolic acid, phosphoenolpyruvate, N-acetylphosphoserine, and phosphoethanolamine cause effective irreversible inhibition of PPase [32, 33]. These compounds are similar to phosphate, the product of the enzymatic reaction, and therefore act as affinity reagents. They bind to the enzyme with the formation of an enzyme-reagent complex and modify the enzyme molecule, thus causing its inhibition. The reaction occurs at the active site, as shown by competitive inhibition of substrate hydrolysis by phosphoric acid monoesters. For this reason, CaPPi, the substrate analog, protects the enzyme from inactivation with phosphoric acid esters. Following complex formation, the phosphorylation of the carboxyl group of a dicarboxylic acid at the active site occurs. The reaction proceeds at low protein concentrations (5-50 µg/ml), which are comparable with cellular concentrations of PPase.
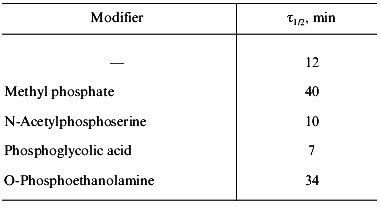
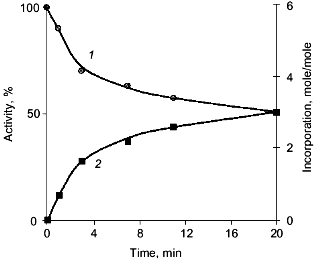
The characteristic feature of PPase interaction with phosphoric acid esters is a clear-cut half-of-the-sites reactivity of subunits. It is manifested by the loss of only 50% of enzymatic activity and incorporation of 3 moles of the reagent into the hexameric molecule (Fig. 9). This behavior is typical both for E- and Y-PPases. Since the enzyme is composed of identical subunits, it was reasonable to suggest that subunit asymmetry results from binding of the reagent to the active site of one subunit and subsequent modification of the structure of the neighboring subunit. To characterize the conformation of the second active half of the molecule, limited proteolysis with subtilisin was used, which was monitored by the rate of inactivation of an unmodified subunit. It appeared that the rate of inactivation depends on the nature of the phosphoric acid monoester associated with the inactive part of the molecule (Table 3).
Table 3. Effect of affinity modification on
E-PPase inactivation by subtilisin
It should be noted that the results and conclusions made above refer to that period of PPase investigations when virtually nothing was known about the spatial structure of these enzymes. However, they agree well with subsequent structure analysis. As shown above, magnesium binding at the active site leads to subunit asymmetry. Sulfate, the phosphate analog, induces the formation of two different trimers, each of which retains the ability of sulfate binding. At the same time, structural changes in PPases induced by interactions with phosphorus-containing analogs of the reaction product are manifested by chemical modification of only half of the enzyme active sites. This poses the problem of active site interactions upon substrate hydrolysis, namely, of the role of quaternary structure in catalysis.Fig. 9. Inactivation of E-PPase by methyl phosphate (1) and incorporation of the reagent into the enzyme molecule (2).
PROPERTIES OF THE E-PPase TRIMER
Are active site interactions preserved during dissociation of a hexamer to trimers? The answer to this question was obtained in 1999. At neutral pH, the equilibrium hexamer <--> trimer is completely biased toward the hexamer. A decrease in pH to 5.0-5.5 causes rapid dissociation of the hexamer. The process is stimulated by decreasing the temperature from 25 to 4°C. However, at low temperatures the dissociation is accompanied by cold inactivation followed by slow reactivation upon returning the protein to room temperature. These experimental details account for variation in estimated dissociation and association constants of E-PPase reported in the literature.
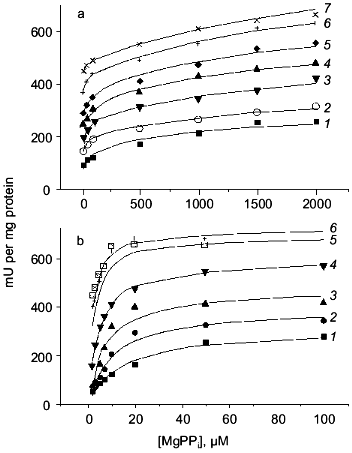
To obtain a trimeric form at neutral pH, two procedures were used. In the first procedure, the enzyme was first kept at slightly acidic conditions; then its concentration was dramatically reduced to prevent association of trimers to hexamers. In the second procedure, low enzyme concentrations were incubated with phosphoethanolamine at pH 7.2; this produced a mixture of two trimers, active and inactive, the latter containing associated phosphoethanolamine. The properties of the trimer were independent of the isolation procedure. Figure 10 shows the dependences of the rate of substrate hydrolysis by E-PPase trimer in a wide range of substrate and Mg2+ concentrations. Parallel experiments with hexameric enzyme are presented for comparison. The results indicate essential differences between the trimer and the hexamer. The reaction catalyzed by a trimer is a two-stage process that cannot be described by Michaelis-Menten kinetics. At the first stage, when the substrate concentration is below 200 µM, half of the maximum rate is achieved. The second stage proceeds at higher substrate concentrations. These results cannot be explained by contamination of trimers with hexamers, since no differences in the kinetics of substrate hydrolysis catalyzed by trimers isolated by different procedures were noted. Furthermore, the formation of hexamers in the presence of PPi and Mg2+ during enzyme activity assay should be excluded, because the accumulation of the product is strictly linear with time. This picture does not change if the enzyme concentration is increased tenfold. Consequently, the observed features of trimer action is characteristic of the trimer. A detailed analysis of several kinetic schemes leads to a single conclusion that the PPase trimer contains at least two substrate binding sites differing in their affinity for substrate with Km values of 3.6 and 985 µM. These data indicate active site interactions in the trimer manifested by negative cooperativity upon substrate binding.
To explain poorer catalytic properties of the trimer in comparison with the hexamer, one could suggest significant conformational changes in trimer subunits caused by disruption of intertrimeric contacts. However, this hypothesis was not supported by experimental data. At the first stage of substrate conversion by the trimer (see Fig. 10), the properties of some trimer subunits and hexamer subunits do not differ significantly (Km = 3.6 and 0.7 µM, respectively). In addition, at substrate concentrations above 1 mM, the catalytic activity of the trimer reaches that of a hexamer. All these findings indicate inconsiderable changes in subunit three-dimensional structure, and even if these changes did occur, their consequences would be similar for all subunits in the trimer. Recall that the properties of trimer and mutant PPases with substitutions of the active site amino acids differ considerably. In both instances, low catalytic efficiency is observed under the conditions when the native PPase exhibits maximum activity. However, at higher substrate concentrations, the activity of the trimer increases and reaches that of the hexamer, while the catalytic activity of mutant proteins remains very low, although their spatial structure, as shown above, is scarcely affected.
Association of the above described trimer with a second trimer obtained by dissociation of one of the mutant proteins yielded chimeric hexamers. All mutants used for this purpose (Asp-70-Asn, Asp-97-Asn, and Lys-104-Gln PPases) showed very low catalytic activity (not more than 1% of that of the native enzyme) but completely retained the ability of binding substrate and Mg2+. It appeared that the properties of the active trimer in such chimeric molecules were virtually identical to those of the trimer in the native hexamer. Therefore, trimer association into the hexamer, even when virtually inactive mutant trimer is used for the second half of the molecule, leads to its activation. Summarizing these data, we conclude that there is cooperation of active sites in the oligomeric PPase structure.Fig. 10. Kinetics of MgPPi hydrolysis by trimeric (a) and hexameric (b) E-PPases. Mg2+ concentration: 0.1 (1), 0.2 (2), 0.5 (3), 1 (4), 2 (5), 5 (6), and 8 mM (7).
The transformation of the non-equivalent active sites of the trimer into identical ones on the association of trimers into hexamers could take place at the stage of substrate or metal ion binding and also on the removal of the products of the reaction. However, since the mutant trimer is capable only of substrate binding without its conversion to products, the transformation of non-equivalent active sites into identical ones in the hexameric molecule is due to the interaction with substrate.
The complex response of PPase to the binding of various ligands (metal ions, substrate, product) indicates the existence of channels of information exchange between active sites of neighboring trimers providing perfect organization and concerted function of the active sites in the catalysis. It seems that these processes include some reorganization of the system of noncovalent interactions in the oligomeric enzyme molecule. It should be emphasized that these informational channels are not identical to the trimer-trimer contacts that are considered critical for maintaining quaternary structure of the protein. This conclusion results, for example, from the properties of His-136-Gln and His-140-Gln E-PPases [29]. These enzymes have higher catalytic activity than the native enzyme in spite of the absence of important trimer-trimer contacts.
The kinetic properties of trimers carrying one or two mutations in the intertrimeric contact zone were reported [29, 34]. In none of the cases were cooperative interactions between trimer subunits shown. The disagreement of these data with our results is probably connected with variation in structure of trimers caused by the amino acid substitutions in the trimers studied in the cited works.
POSSIBLE REGULATION MECHANISMS OF PPase ACTIVITY
PPases are constitutive enzymes; therefore, the regulation of their catalytic activity should occur at the synthesized-protein level. These processes are largely unknown, but several mechanisms are possible. One of them suggesting Ca2+-mediated activity regulation has been described in [20] of this volume.
Another regulation mechanism is based on the interactions of PPases with phosphoric acid monoesters, abundant phosphorus-containing cellular components. These compounds effectively modify PPases, decreasing their activity by half. It is known that the acylphosphate bond is labile. In the presence of PPi and imidodiphosphate, the enzyme activity is rapidly restored due to acylphosphate hydrolysis and elimination of the reagent. These facts provide evidence for the involvement of monoesters in regulation of PPase activity.
The ability of phosphoethanolamine to completely shift hexamer-trimer equilibrium toward the trimer at neutral pH deserves special attention. Since as shown above, the catalytic efficiency of a trimer is lower than that of a hexamer, enzyme dissociation may be a mechanism controlling enzymatic activity. In this connection, a detailed study of a wide range of natural phosphoric acid esters capable of modifying PPase oligomeric structure is now in progress.
The author's works described in this review were supported in part by the Russian Foundation for Basic Research (grants No. 97-04-50031 and No. 96-15-97969) and the “Universities of Russia” Program (basic investigations, grant No. 5077).
REFERENCES
1.Shintani, T., Uchiumi, T., Yonezawa, T., Salminen,
A., Baykov, A., Lahti, R., and Hachimori, A. (1998) FEBS Lett.,
439, 263-266.
2.Harutyunyan, E. H., Terzyan, S. S., Voronova, A.
A., Kuranova, I. P., Smirnova, E. A., Vainshtein, B. E., Hohne, W. E.,
and Hansen, G. (1981) Dokl. Akad. Nauk SSSR, 8,
1481-1485.
3.Oganessyan, V. Yu., Kurilova, S. A., Vorobyeva, N.
N., Nazarova, T. I., Popov, A. N., Lebedev, A. A., Avaeva, S. M., and
Harutyunyan, E. G. (1994) FEBS Lett., 348, 301-304.
4.Kankare, J., Neal, G., Salminen, T., Glumoff, T.,
Cooperman, B., Lahti, R., and Goldman, A. (1994) Protein Eng.,
7, 823-830.
5.Kankare, J., Salminen, T., Lahti, R., Cooperman,
B., Baykov, A., and Goldman, A. (1996) Acta Crystallogr.,
D52, 551-563.
6.Teplyakov, A., Obmolova, G., Wilson, K. S., Ishii,
K., Kaji, H., Samejima, T., and Kuranova, I. (1994) Protein
Sci., 3, 1098-1107.
7.Leppanen, V.-M., Nummelin, H., Hansen, T., Lahti,
R., Schafer, G., and Goldman, A. (1999) Protein Sci., 8,
1218-1231.
8.Harutyunyan, E. G., Oganessyan, V. Yu., Oganessyan,
N. N., Terzyan, S. S., Popov, A. N., Rubinskiy, S. V., Vainshtein, B.
K., Nazarova, T. I., Kurilova, C. A., Vorobyeva, N. N., and Avaeva, S.
M. (1996) Kristallografiya, 41, 84-96.
9.Kankare, J., Salminen, T., Lahti, R., Cooperman,
B., Baykov, A., and Goldman, A. (1996) Biochemistry, 35,
4670-4677.
10.Harutyunyan, E. H., Oganessyan, V. Yu.,
Oganessyan, N. N., Avaeva, S. M., Nazarova, T. I., Vorobyeva, N. N.,
Kurilova, S. A., Huber, R., and Mather, T. (1997) Biochemistry,
36, 7754-7760.
11.Avaeva, S. M., Rodina, E. V., Vorobyeva, N. N.,
Kurilova, C. A., Nazarova, T. I., Sklyankina, V. A., Oganessyan, V.
Yu., and Harutyunyan, E. H. (1998) Biochemistry
(Moscow),63, 592-599.
12.Avaeva, S., Kurilova, S., Nazarova, T., Rodina,
E., Vorobyeva, N., Sklyankina, V., Grigorjeva, O., Harutyunyan, E.,
Oganessyan, V., Wilson, K., Dauter, Z., Huber, R., and Mather, T.
(1997) FEBS Lett., 410, 502-508.
13.Harutyunyan, E. H., Kuranova, I. P., Vainstein,
B. K., Hohne, W. E., Lamzin, V. E., Dauter, Z., Teplyakov, A. V., and
Wilson, K. S. (1996) Eur. J. Biochem., 239, 220-228.
14.Heikinheimo, P., Lehtonen, J., Baykov, A., Lahti,
R., Cooperman, B., and Goldman, A. (1996) Structure, 4,
1491-1508.
15.Avaeva, S. M., Rodina, E. V., Vorobyeva, N. N.,
Kurilova, S. A., Nazarova, T. I., Sklyankina, V. A., Oganessyan, V.
Yu., Samygina, V. R., and Harutyunyan, E. H. (1998) Biochemistry
(Moscow), 63, 671-684.
16.Tuominen, V., Heikinheimo, P., Kajander, T.,
Torkkel, T., Hyytia, T., Kapyla, J., Lahti, R., Cooperman, B., and
Goldman, A. (1998) J. Mol. Biol., 284, 1565-1580.
17.Shafranskii, Yu. A., Baykov, A. A., Andrukovich,
P. F., and Avaeva, S. M. (1977) Biokhimiya, 42,
1244-1251.
18.Schlesinger, M., and Coon, M. (1960) Biochim.
Biophys. Acta, 41, 30-36.
19.Mel'nik, M. S., Nazarova, T. I., and Avaeva, S.
M. (1982) Biokhimiya, 47, 323-328.
20.Avaeva, S. M., Vorobyeva, N. N., Kurilova, S. A.,
Nazarova, T. I., Polyakov, K. M., Rodina, E. V., and Samygina, V. R.
(2000) Biochemistry (Moscow), 65, 373-387.
21.Borschik, I. B., Magretova, N. N., Cherniyak, V.
Ya., Sklyankina, V. A., and Avaeva, S. M. (1986) Biokhimiya,
51, 1484-1489.
22.Salminen, T., Kapyla, J., Heikinheimo, P.,
Kankare, J., Goldman, A., Heinonen, J., Baykov, A., Cooperman, B., and
Lahti, R. (1995) Biochemistry, 34, 782-791.
23.Avaeva, S., Ignatov, P., Kurilova, S., Nazarova,
T., Rodina, E., Vorobyeva, N., Oganessyan, V., and Harutyunyan, E.
(1996) FEBS Lett., 399, 99-102.
24.Heikinheimo, P., Pohjanjoki, P., Helminen, A.,
Tasanen, M., Cooperman, B., Goldman, A., Baykov, A., and Lahti, R.
(1996) Eur. J. Biochem., 239, 138-143.
25.Avaeva, S., Rodina, E., Kurilova, S., Nazarova,
T., and Vorobyeva, N. (1996) FEBS Lett., 392, 91-94.
26.Pohjanjoki, P., Lahti, R., Goldman, A., and
Cooperman, B. (1998) Biochemistry, 37, 1754-1761.
27.Kapyla, J., Hyytia, T., Lahti, R., Goldman, A.,
Baykov, A., and Cooperman, B. (1995) Biochemistry, 34,
792-800.
28.Fabrichny, I., Kasho, V., Hyytia, T., Salminen,
T., Halonen, P., Dudarenkov, V., Heikinheimo, P., Chernyak, V.,
Goldman, A., Lahti, R., Cooperman, B., and Baykov, A. (1997)
Biochemistry, 36, 7746-7753.
29.Baykov, A., Dudarenkov, V., Kapyla, J., Salminen,
T., Hyytia, T., Kasho, V., Husgafvel, S., Cooperman, B., and Lahti, R.
(1995) J. Biol. Chem., 270, 30804-30812.
30.Baykov, A., Tam-Villoslado, J., and Avaeva, S.
(1979) Biochim. Biophys. Acta, 569, 228-238.
31.Avaeva, S. M., Vorobyeva, N. N., Mel'nik, M. S.,
and Nazarova, T. I. (1979) Bioorg. Khim., 5,
1570-1577.
32.Svyato, I., Nazarova, T., Sklyankina, V., and
Avaeva, S. (1984) FEBS Lett., 167, 269-272.
33.Sklyankina, V., and Avaeva, S. (1990) Eur. J.
Biochem., 191, 195-201.
34.Velichko, I., Mikalahti, K., Kasho, V.,
Dudarenkov, V., Hyytia, T., Goldman, A., Cooperman, B., Lahti, R., and
Baykov, A. (1998) Biochemistry, 37, 734-740.