REVIEW: Inorganic Polyphosphate and Polyphosphate Kinase: Their Novel Biological Functions and Applications
T. Shiba1*, K. Tsutsumi1, K. Ishige2, and T. Noguchi2
1Division of Molecular Chemistry, Graduate School of Engineering, Hokkaido University, Sapporo 060-8628, Japan; fax: +81-11-706-7816; E-mail: shiba@dove-mc.eng.hokudai.ac.jp2Biochemical Divisions, YAMASA Corporation, Choshi, Chiba 288-0056, Japan
* To whom correspondence should be addressed.
Received October 4, 1999
In this review, we discuss the following two subjects: 1) the physiological function of polyphosphate (poly(P)) as a regulatory factor for gene expression in Escherichia coli, and 2) novel functions of E. coli polyphosphate kinase (PPK) and their applications. With regard to the first subject, it has been shown that E. coli cells in which yeast exopolyphosphatase (poly(P)ase), PPX1, was overproduced reduced resistance to H2O2 and heat shock as did a mutant whose polyphosphate kinase gene is disrupted. Sensitivity to H2O2 and heat shock evinced by cells that overproduce PPX1 is attributed to depressed levels of rpoS expression. Since rpoS is a central element in a regulatory network that governs the expression of stationary-phase-induced genes, poly(P) affects the expression of many genes through controlling rpoS expression. Furthermore, poly(P) is also involved in expression of other stress-inducible genes that are not directly regulated by rpoS. The second subject includes the application of novel functions of PPK for nucleoside triphosphate (NTP) regeneration. Recently E. coli PPK has been found to catalyze the kination of not only ADP but also other nucleoside diphosphates using poly(P) as a phospho-donor, yielding NTPs. This nucleoside diphosphate kinase-like activity of PPK was confirmed to be available for NTP regeneration essential for enzymatic oligosaccharide synthesis using the sugar nucleotide cycling method. PPK has also been found to express a poly(P):AMP phosphotransferase activity by coupling with adenylate kinase (ADK) in E. coli. The ATP-regeneration system consisting of ADK, PPK, and poly(P) was shown to be promising for practical utilization of poly(P) as ATP substitute.
KEY WORDS: polyphosphate, polyphosphate kinase, exopolyphosphatase, rpoS, stress-inducible genes, NTP regeneration, nucleoside diphosphate kinase, adenylate kinase, glycosyltransferase, oligosaccharides, N-acetyllactosamine, poly(P):AMP phosphotransferase
Abbreviations: ADK) adenylate kinase; NDP) nucleoside diphosphates; NTP) nucleoside triphosphates; PAP) polyphosphate:AMP phosphotransferase; PPK) polyphosphate kinase; PPX1) yeast exopolyphosphatase.
Inorganic polyphosphate (poly(P)) has been found to be widely
distributed in every living thing so far examined, from bacteria to
mammals. Several biological functions have been suggested for cellular
poly(P), including a reservoir for phosphate and energy, a chelator of
metal ions, a buffer against alkali, and a channel for DNA entry [1]. However, the details of these poly(P) functions
are still uncertain.
Several enzymes that contribute to the metabolism of poly(P) including polyphosphate kinase (PPK), exopoly(P)ase, poly(P):AMP phosphotransferase (PAP), polyphosphate glucokinase, and endopolyphosphatase have been identified [1]. PPK that is responsible for the synthesis of poly(P) polymerizing terminal phosphate of ATP to a poly(P) in a freely reversible reaction [1-5] has been most intensively studied.
Here, we describe novel biological functions of poly(P) and PPK in Escherichia coli and their applications.
1. Poly(P)-DEPENDENT GENE EXPRESSION IN E. coli
1.1. PPX1-overproducing cells show no cellular poly(P) accumulation and the same characteristics as those of a ppk mutant. An E. coli ppk mutant is deficient in functions expressed in the stationary phase and fails to survive [6]. The mutant also showed a lack of resistance to an oxidant (H2O2), to a redox-cycling agent (menadione), to heat, and to an osmotic challenge [6, 7]. In stationary culture of the ppk mutant, small colony variants with improved viability and resistance to H2O2 frequently appeared [7]. Although survivors of the ppk mutant in the stationary phase are far fewer in number after a week or so, the mutants are replaced by an outgrowth of a small-colony-size variant with a stable genotype and with improved viability and resistance to H2O2 and heat [7]. Since neither PPK activity nor long-chain poly(P) was restored in the small-colony variant [7], unknown mutations that suppress the poly(P)-minus phenotype might occur during cultivation of the ppk mutant.
To overcome these unstable features of the ppk mutant, we employed the plasmid system that overproduces recombinant yeast exopolyphosphatase (PPX1) [8] to remove as much of the cellular poly(P) as possible. In this system, there are the following two advantages for examining the poly(P)-minus phenotype. First, it is possible to avoid the appearance of the small-colony-size variant by using a fresh transformant carrying the PPX1 gene on plasmids, and it is also possible to observe the poly(P)-minus features with good reproducibility. Second, it may be possible to remove short-chain poly(P), which suppresses the poly(P)-minus effect, because the ppk mutant still makes short poly(P) chains of about 60 residues in size by an alternative synthetic pathway [9]. Based on these advantages, we examined the mechanism underlying the involvement of poly(P) in H2O2 sensitivity.
To confirm that PPX1 overproduction can efficiently remove cellular poly(P), KT1008 cells (F-,Delta(arg-lac)U169, araD139, rpsL150, ptsF25, flbB5301, rbsR, deoC1) were transformed by either a plasmid that carries the PPX1 gene under the trc promoter (pTrcPPX1) or an original vector of pTrcPPX1 (pTrcHisB) [10]. Cells were cultured to the mid-logarithmic growth phase (OD = 0.4) in LB and starved of nitrogen for 4 h in Mops minimal medium. Cellular poly(P) rapidly accumulated within 30 min after starvation and gradually decreased thereafter in pTrcHisB transformants, whereas no poly(P) accumulation was observed in pTrcPPX1 transformants [10]. This result clearly showed that PPX1 overproduction efficiently removes cellular poly(P).
It was also confirmed that pTrcPPX1 transformants have the same features as those of the ppk mutant. H2O2 sensitivity of pTrcPPX1 transformants was almost 10-fold greater than that of control vector (pTrcHisB) transformants in the wild type. Also, the heat shock sensitivity of pTrcPPX1 transformants was much higher than that of the control vector transformants (Fig. 1a). These characteristics were the same as those of the phenotype of the ppk mutant [7]. Moreover, introduction of extra copies of the ppk gene by multiple-copy plasmid into PPX1-overproducing cells can overcome this high sensitivity to H2O2, almost restoring them to the levels of vector transformants [10]. These results suggested that the high sensitivity to these stresses caused by PPX1 overproduction is dependent on the poly(P) level in cells.
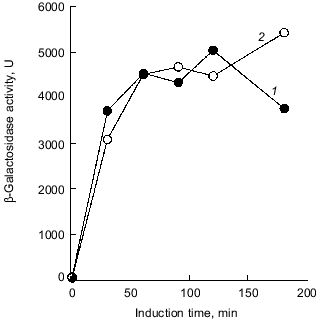
Intriguingly, the H2O2 sensitivity of pTrcPPX1 transformants was approximately 1,000-fold greater than that of vector transformants in the katG mutant, whereas, in the katE mutant, the sensitivity of pTrcPPX1 transformants were only 10-fold greater than that of vector transformants like wild type cells [10]. This means that the high sensitivity of pTrcPPX1 transformants is caused mainly by the deficiency of the katE gene products, HPII, since there are only two catalase activities--KatG (HPI) and KatE (HPII)--in E. coli.Fig. 1. a) Effect of PPX1 overproduction on heat sensitivity. The wild-type strain KT1008 harbored either the plasmid with the PPX1 gene (pTrcPPX1) (1) or the control vector (pTrcHisB) (2). After cells were incubated at 55°C, the viable cell number was determined by plating onto LB agar. b) Expression of the katE-lacZ operon fusion monitored by beta-galactosidase activity [32]. KT1008EL cells (as KT1008 but lambdaRS45 : katE-lacZ) [33] harbored a plasmid or pairs of plasmids as follows: PPX1 gene (pTrcPPX1) (1), control vector for PPX1 (pTrcHisB) (2), a pair of pTrcPPX1 and pBS-rpoS (carrying the rpoS gene) (3) [10] or a pair of pTrcHisB and pBS-rpoS (4). KatE expression was induced by nitrogen starvation with Mops minimal medium [10].
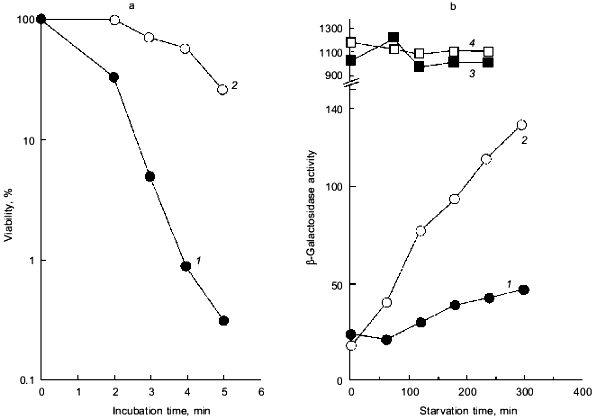
1.2. Poly(P) affects rpoS expression. Deficiency of KatE activity in PPX1-overproducing cells resulted in a low-level induction of katE expression as was the case with the ppk mutant (Fig. 1b) [10]. When extra copies of the rpoS gene on the multiple copy plasmid (pBS-rpoS) [10] were introduced into a PPX1 overproducing cell, katE transcription was fully induced (Fig. 1b). Since the expression of katE was found to be dependent on rpoS, the high sensitivity to H2O2 in pTrcPPX1 transformants is attributed to the deficiency of RpoS function. The product of rpoS is the sigma38 subunit of RNA polymerase responsible for the expression of nearly 50 genes, including katE, involved in adjustments in the stationary phase to nutrient deprivations, high osmolarity, and other stressful agents such as H2O2 [11]. During the course of analysis of the ppk mutant, high sensitivity to heat shock in this mutant was recovered by introduction of multiple-copy rpoS plasmids [7]. These results indicate not only that the characteristics of PPX1-overproducing cells are equivalent to those of the ppk mutant, but the high sensitivity to H2O2, and heat shock in the ppk mutant resulted in the deficiency of rpoS expression.
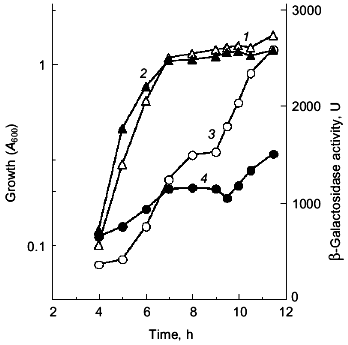
Since the poly(P)-minus phenotype was attributed to RpoS function, poly(P) somehow regulates rpoS expression at the level of transcription, translation, and/or stabilization of the sigma38 protein post-translationally. First, the effect of poly(P) on rpoS transcription was examined using a lysogen carrying the rpoS-lacZ transcriptional fusion gene. KT1008SL (as KT1008 but lambdaRS45 : rpoS-lacZ), which was lysogenyzed by lambda_carrying the rpoS-lacZ transcriptional fusion gene, harboring either pTrcPPX1 or pTrcHisB (vector) was grown in LB medium and rpoS transcription was monitored along the growth curve (Fig. 2) [10]. Vector transformants exhibited induction during entry into the stationary phase, and the transcription increased by about 5-fold after 12 h of cultivation. In the pTrcPPX1 transformants, the transcription increased by only 3-fold in stationary phase (12 h of cultivation). These results suggest that the induction of rpoS transcription is dependent on cellular poly(P).
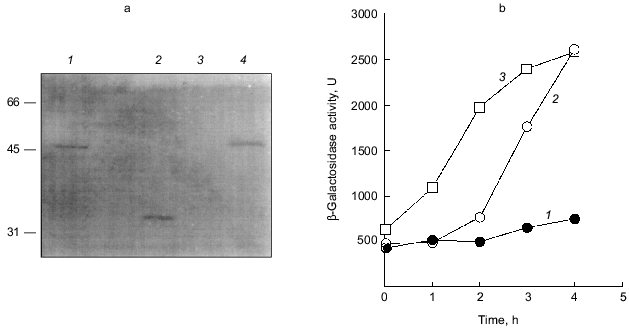
The overproduction of certain proteins may have unknown effects on such physiological activities of cells as growth, transcription, and translation. To rule out the possibility that PPX1 overproduction itself inhibits gene expression nonspecifically, an inactive PPX1 gene was overproduced and the effect of the overproduction on rpoS transcription was examined. pTrcDeltaPPX1, which carries a truncated PPX1 gene encoding a PPX1 deletion (DPPX1) protein, was introduced into KT1008SL. Poly(P)ase activity of DPPX1 was not detected (data not shown) and both DPPX1 and PPX1 were shown by Western blotting to be overproduced at the same levels (Fig. 3a, lanes 2 and 4). The levels of induction of rpoS expression in the cells harboring these plasmids were then compared (Fig. 3b). Expression of rpoS was induced both in the vector-only transformants and DPPX1-overproducing cells, whereas no induction was observed in pTrcPPX1 transformants. The overproduction of inactive PPX1 did not inhibit the induction of rpoS expression, indicating that the low-level induction of rpoS expression in pTrcPPX1 transformants is attributed to poly(P)ase activity, not to an artifact of protein overproduction.Fig. 2. Expression by the rpoS-lacZ operon fusion during growth in LB [10]. KT1008 cells harbored either plasmids with the PPX1 gene (pTrcPPX1) (2, 4) or the control vector (pTrcHisB) (1, 3). 1, 2) Growth; 3, 4) beta-galactosidase activity.
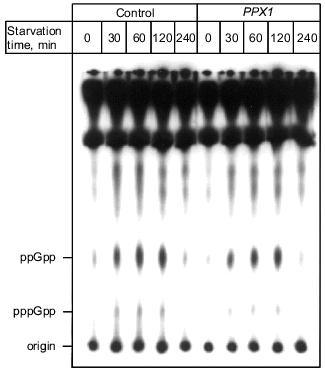
1.3. PPX1 overproduction does not affect (p)ppGpp accumulation. (p)ppGpp are positive signals leading to the accumulation of sigma38 [12, 13]. If poly(P) controls the cellular levels of (p)ppGpp, the level of rpoS expression is also altered depending on poly(P). To estimate the cellular levels of (p)ppGpp, they were extracted from CF3376 (DeltagppA) [14], which is deficient in pppGpp hydrolyase (a gppA gene product). For ppGpp alanysis, it is possible to detect pppGpp much more efficiently using the gppA mutant strain rather than the wild type. As shown in Fig. 4, both ppGpp and pppGpp accumulated after 30 min of nitrogen starvation by Mops minimal medium, and they stayed at the same level for 2 h. Accumulation was observed both in pTrcPPX1 and pTrcHisB transformants. No effect of PPX1 overproduction was observed on (p)ppGpp accumulation, suggesting that the reduced level of poly(P) by prior PPX1 overproduction does not affect the cellular (p)ppGpp level.Fig. 3. Effect of DPPX1 overproduction on rpoS expression. a) Expression level of PPX1 and DPPX1. Aliquots of the crude extracts containing 8.5 µg of protein were separated by 12.5% SDS-PAGE, and the proteins were visualized by Western blotting with the alkaline phosphatase conjugate Ni-NTA for detection of the His-tagged PPX1 protein. 1) Purified PPX1 protein; 2) KT1008/pTrcDeltaPPX1 extract; 3) KT1008/pTrcHisB extract; 4) KT1008/pTrcPPX1 extract. On the left, molecular masses of protein markers in kD. b) Induction of rpoS expression by nitrogen starvation. RpoS-lacZ expression was monitored in KT1008 cells harboring the following plasmids: pTrcPPX1 (1), pTrcHisB (2), and pTrcDeltaPPX1 (3).
1.4. Involvement of poly(P) in SOS regulated genes. Since poly(P) plays a role in the regulation of rpoS expression, the possibility of involvement of poly(P) in other stress-inducible genes was examined. First, poly(P) dependence of expression of the major DNA damage-inducible genes, which are elements of the SOS regulon, were analyzed, because resistance to mitomycin C (MMC) and UV was decreased in cells in which PPX1 was overproduced [15]. Induction of umuDC expression, which is promoted by inactivation of the LexA repressor by activated RecA, was also greatly diminished in PPX1-overproducing cells when the cells were exposed by MMC. Under the same condition, recA expression was also diminished and not efficiently induced by MMC exposure [15]. Diminished expression of umuDC and recA might be due to the low-level induction of recA itself. Also, diminished expression of recA means that expression of all genes involved in the SOS regulon is not induced efficiently. However, the RecA protein was almost fully activated both in control and PPX1-overproducing cells because the proteolytic cleavage of UmuD, which was induced by activated-RecA protein, did not depend on poly(P) [15]. Based on these findings, poly(P) may directly affect induction of the expression of recA, and may be necessary for new synthesis of recA mRNA. This is consistent with the results of poly(P)-dependent expression (transcription induction) of rpoS. Poly(P) somehow plays a crucial role in the induction of expression on stress-inducible genes through regulation of rpoS and recA transcription.Fig. 4. Effect of PPX1 overproduction on (p)ppGpp accumulation. At the times indicated, starved cells were collected, and (p)ppGpp was extracted [34]. (p)ppGpp spots were separated by TLC and identified by authentic (p)ppGpp as standards [34].
To rule out the possibility that overproduction of PPX1 diminishes overall transcription efficiency, the effect of poly(P) on induction of gene expression under the lacUV5 promoter by isopropyl beta-D-thiogalactopyranoside (IPTG) was examined. KT1008 cells harboring a plasmid with a lacUV5-lacZ fusion gene (pUR290) [16] and either a plasmid with the PPX1 or the control vector were used for monitoring promoter activity of lacUV5. No significant difference in transcription efficiency between vector-only transformants and PPX1 transformants was observed (Fig. 5). Thus gene expression under the lacUV5 promoter was not dependent on the cellular poly(P) level.
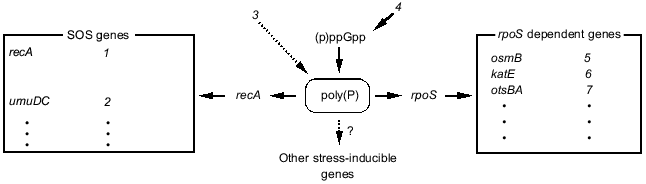
1.5. Hypothetical model of poly(P) functions on gene expression regulation. Poly(P)-dependent regulation of gene expression is summarized in Fig. 6. Poly(P) controls rpoS and recA expression at the transcription level and affects the expression of many stress-inducible and stationary phase-inducible genes. Poly(P) did not have any effect on (p)ppGpp accumulation. On the other hand, (p)ppGpp accumulation is necessary for accumulation of cellular poly(P) [17, 18]. (p)ppGpp may be an upstream signal and poly(P) is an intermediate signal for induction of rpoS expression. However, the correlation between (p)ppGpp accumulation and recA expression is still difficult to explain because there is no report of (p)ppGpp accumulation being required for induction of SOS genes. Further analyses are needed to elucidate the detailed mechanism of the regulation of recA expression by poly(P).Fig. 5. Expression of the lacUV5-lacZ operon fusion monitored by beta-galactosidase activity. KT1008 cells harboring a plasmid with a lacUV5-lacZ fusion gene and either a plasmid with the PPX1 gene (pSUPPX1) [10] (1) or the control vector (pSUHisB) [10] (2) were cultured to the mid-logarithmic phase, and IPTG was added to the final concentration of 1 mM. At the times indicated, cells were collected and beta-galactosidase activity was measured [32].
Kusano and Ishihama [19] reported that poly(P) physically interacts with RNA polymerase and inhibits transcription in vitro by RNA polymerase containing sigma70. Since poly(P) rapidly accumulates just after stress and starvation, and then gradually decreases [10, 15, 18, 20], it may be a signal molecule that changes the cellular state, like (p)ppGpp [12] and cAMP. Poly(P) may directly affect the transcription machinery by modifying RNA polymerase. It is also possible that poly(P) affects the stability of mRNA, since Blum et al. [21] suggested that poly(P) and PPK interact with RNA degradosome. Although the precise mechanism is still unclear, it is thought that poly(P) is closely related to RNA metabolism in E. coli cells.Fig. 6. Schematic model for gene expression regulation by poly(P). rpoS regulated genes and SOS genes are described in [35, 36]. Dotted lines with an arrow represent the hypothetical connection. SOS genes: 1) activation of expression of SOS genes, recombination; 2) error-prone repair; 3) DNA damage; 4) starvation. rpoS dependent genes: 5) osmotic stress; 6) oxidative stress; 7) thermotolerance.
2. NOVEL FUNCTIONS OF POLYPHOSPHATE KINASE AND THEIR
APPLICATIONS
2.1. Possible involvement of poly(P) and PPK in nucleotide metabolism. PPK has been known to catalyze the reversible transfer of a phosphate molecule between poly(P) and ATP [1-5]. Recently, Kuroda and Kornberg [22], and we [23] independently found that E. coli PPK can catalyze the transfer of a phosphate molecule from poly(P) to another nucleoside diphosphate (NDP), yielding a nucleoside triphosphate (NTP). Thus, PPK may provide a nucleoside diphosphate kinase (NDK)-like activity as follows:
ADP + poly(P)n <--> ATP + poly(P)n-1,
NDP + poly(P)n -->_NTP + poly(P)n-1 (NTP: GTP, CTP, UTP).
Unlike NDK, the reaction catalyzed by PPK is not reversible except in the case of ADP, and PPK cannot catalyze the complete kination of NDP even in the presence of excess poly(P) [23]. Kuroda and Kornberg reported that the kination efficiency of NDP substrates was as follows: ADP > GDP > UDP > CDP [22]. It is obvious that PPK prefers purine nucleotides to pyrimidine nucleotides. In addition, PPK extracted from Pseudomonas aeruginosa cells was also reported to have the same activity and preference in substrates, suggesting that this property is a general feature of PPK [22]. Although the biological significance of the NDK-like activity of PPK and its substrate specificity is not clear, poly(P) may be able to serve as a direct phospho-donor for the kination of not only ADP but also other NDPs by the action of PPK.
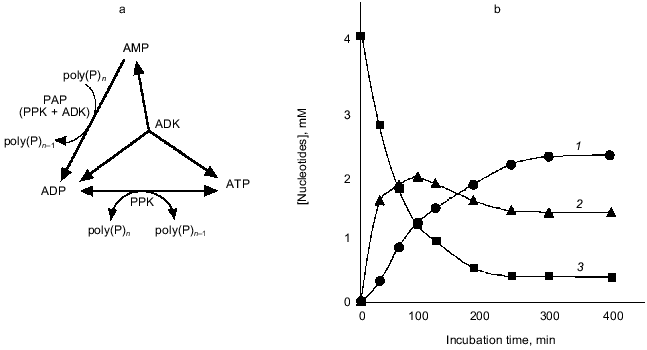
Compared to PPK, PAP, which catalyzes the reversible transfer of a phosphate residue from poly(P) to AMP yielding ADP, has not been studied in detail. Marked PAP activity was found in Acinetobacter johnsonii [24]and Myxococcus xanthus [1], and weak PAP activity was also detected in E. coli cells [1]. We found that the overexpression of E. coli ppk gene encoding PPK results in a drastic increase in PAP activity, although the PPK itself has no detectable PAP activity [25]. Therefore, we investigated the mechanism of enhanced expression of PAP activity by overproduction of PPK in E. coli cells, and we confirmed that PPK requires adenylate kinase (ADK) to express PAP activity [25]. Although PPK and ADK synergistically constitute PAP activity, this novel activity cannot be explained by the mere combination of the authentic activities of both enzymes. The mechanism of the constitution of PAP activity is speculated to be as follows: under PAP reaction conditions, PPK phosphorylates itself using poly(P) as a phospho-donor but cannot catalyze the phosphate transfer reaction because of the absence of a phospho-acceptor. On the contrary, ADK has a phospho-acceptor (AMP) but not a phospho-donor. When both enzymes exist together, phosphorylated PPK may provide its phosphate molecule to ADK followed by the phosphate-transfer reaction to AMP catalyzed by ADK, yielding ADP.
As shown in Fig. 7, in the presence of poly(P), AMP was immediately converted to ADP by PAP activity created by PPK and ADK, and the formed ADP was next converted to ATP due to a reversible poly(P):ADP phosphotransferase activity of PPK and an authentic activity of ADK that catalyzes the reversible phosphorylation of ADP using another ADP as a phospho-donor [26]. After a while, phosphorylation reactions seemed to cease, and for a while the concentration of each adenine nucleotide remained at the same level, suggesting that PPK and ADK cooperate to maintain the levels of each adenine nucleotide and poly(P). ADK is responsible for the maintenance of an appropriate energy state of the cell [26] and functions as NDP kinase to generate NTP from NDP using ATP as a phospho-donor [27]. PPK has been believed to be responsible for utilization of high energy of poly(P) by catalyzing the generation of ATP from ADP [1]. It is of particular interest that ADK can indirectly utilize poly(P) as a phospho-donor instead of ATP in the presence of PPK, and PPK itself can serve as a poly(P)-dependent NDK. It is most likely that ADK and PPK compensate each other to metabolize nucleotides upon the intracellular level of ATP and poly(P). The novel functions of E. coli PPK described here reveal that PPK is possibly involved in the maintenance of the cell energy state by not only catalyzing the phosphate-transfer reaction between ATP and poly(P) but also metabolizing nucleotides with other nucleotide-metabolizing enzymes directly using the high energy of poly(P).
2.2. Applications of poly(P) as an ATP substitute and energy source. PPK's ability for phosphorylation of ADP using poly(P) as a phospho-donor has been shown to be considerably useful for an ATP-regeneration for the enzymatic reaction consuming ATP as a phospho-donor or an energy source, compared to the expensive ATP regeneration systems that have been used in laboratories, such as the combination of creatine kinase and phosphocreatine [1, 28]. However, it has not been easy to obtain a sufficient amount of PPK from bacterial cells because the cells have small amounts of PPK but are rich in ATP-degrading enzymes. Accordingly the combination of PPK and poly(P) has not been utilized for ATP-regeneration even for laboratory use.Fig. 7. Formation of ATP and ADP from AMP by PPK and ADK. a) Diagram of ATP generation from AMP by PPK and ADK. AMP is converted to ADP by PAP activity consisting of PPK and ADK. ATP was formed from ADP by authentic activities of both PPK and ADK. b) Conversion of AMP to ADP and ATP. The reaction mixture contained 100 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 100 mM (NH4)2SO4, 4.0 mM AMP, poly(P) (75 mM as phosphate), 0.1 U/ml PPK, and 0.25 U/ml ADK and was incubated at 37°C. 1, 2, 3) ATP, ADP, and AMP, respectively.
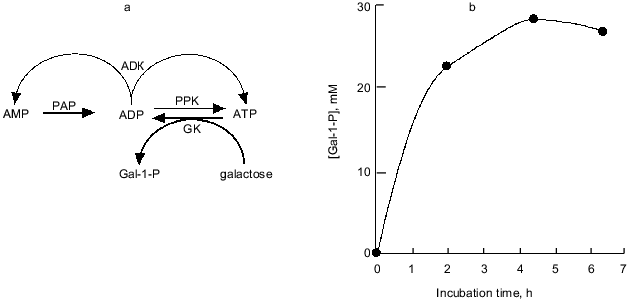
Nowadays, the overproduction of PPK achieved by recombinant DNA technology enables the practical use of PPK and poly(P). We synthesized galactose 1-phosphate (Gal-1-P) with E. coli galactokinase and an ATP-regeneration system consisting of poly(P) and PPK prepared from PPK-overproducing E. coli cells. The phosphorylation efficiency of galactose with poly(P) and PPK was shown to be almost the same as that with ATP (data not shown). As described before, with ADK, PPK catalyzes the formation of ADP from AMP, followed by ATP formation from ADP by authentic activities of both enzymes. Thus, the addition of ADK to an ATP-regeneration system consisting of poly(P) and PPK is more promising because cheap AMP can substitute for expensive ATP or ADP as an essential compound initially added to the reaction system, and the supplement of ADK significantly enhances the efficiency of the ATP-regeneration possibly because ADK can efficiently catalyze the phosphorylation of ADP using another ADP as a phospho-donor yielding ATP and AMP [26]. Eventually, using only 4 mM AMP, 28 mM Gal-1-P was synthesized by the action of PPK and ADK in the presence of poly(P), as shown in Fig. 8.
Recently biologically active oligosaccharides have been enzymatically synthesized using sugar nucleotides and glycosyltransferases. Due to the expense of sugar nucleotides, however, the enzymatic synthesis had not been thought to be suitable for practical production of oligosaccharides. Therefore, Wong et al. developed the sugar nucleotide cycling (SNC) technology, in which sugar nucleotides are enzymatically regenerated from sugar and NTP [29-31]. Since NDPs generated through the glycosyltransferase reaction should be converted to NTP, Wong et al. used pyruvate kinase and phosphoenol pyruvate (PEP) as phospho-donors for NTP-regeneration. The problem in their system is that PEP is expensive and not readily available. As described in the previous section, PPK has been found to be able to phosphorylate NDPs to give NTPs using poly(P) as a phospho-donor. Therefore, we examined the possibility of using poly(P) and PPK in place of PEP and pyruvate kinase for enzymatic oligosaccharide synthesis using SNC technology, because poly(P) is quite cheap compared to PEP.Fig. 8. Synthesis of galactose 1-phosphate (Gal-1-P) with PPK and ADK. a) Diagram of galactose 1-phosphate synthesis with ATP-regeneration catalyzed by PPK and ADK. AMP (4 mM) and ADP are phosphorylated by PAP and authentic activities of PPK and ADK, respectively. Galactokinase (GK) catalyzes the formation of Gal-1-P using ATP as a phospho-donor. ADP should be regenerated to ATP by PPK and ADK. b) Synthesis of Gal-1-P with ATP-regeneration using AMP as a primary substrate. The reaction mixture contained 100 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 4.0 mM AMP, poly(P) (75 mM as phosphate), 40 mM galactose, 0.2 U/ml galactokinase (E. coli), 0.1 U/ml PPK, and 0.25 U/ml ADK and was incubated at 37°C.
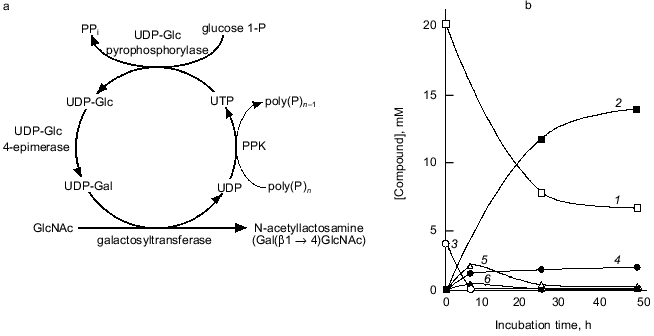
We attempted to synthesize N-acetyllactosamine (Gal(beta1-4)GlcNAc) using the NDK-like activity of PPK, as shown in Fig. 9a. UDP-Glc pyrophosphorylase and UDP-Glc-4-epimerase catalyze the synthesis of UDP-Glc from glucose 1-phosphate and UTP, and the isomerization of UDP-Glc to UDP-Gal, respectively. The galactosyltransferase transfers galactose from UDP-Gal to N-acetylglucosamine, yielding N-acetyllactosamine. UDP formed through the galactosyltransferase reaction should be regenerated to UTP by the action of PPK in the presence of poly(P). As shown in Fig. 9b, 14 mM N-acetyllactosamine was accumulated after 48 h of reaction even though only 4 mM UTP was initially added, demonstrating that PPK and poly(P) efficiently catalyze the regeneration of UTP from UDP [23]. It is obvious that poly(P) and PPK can replace PEP and pyruvate kinase in regeneration of NTPs and are available for enzymatic cyclic synthesis of oligosaccharides.
Fig. 9. Cyclic synthesis of N-acetyllactosamine with UTP-regeneration by PPK. a) Representative of system for cyclic synthesis of N-acetyllactosamine. Abbreviations: UDP-Glc) UDP-glucose; UDP-Gal) UDP-galactose; glucose 1-P) glucose 1-phosphate; GlcNAc) N-acetylglucosamine. b) Synthesis of N-acetyllactosamine with PPK. The reaction mixture contained 150 mM Tris-HCl (pH 7.8), 10 mM MgCl2, 10 mM MnCl2, 20 mM N-acetylglucosamine, 4 mM UTP, 30 mM glucose 1-phosphate, poly(P) (75 mM as phosphate), 0.1 U/ml PPK, 1.0 U/ml UDP-Glc pyrophosphorylase, 1.0 U/ml UDP-Glc 4-epimerase, and 0.5 U/ml galactosyltransferase (from bovine milk, Sigma, USA), and was incubated at 37°C. 1) GlcNAc; 2) N-acetylglucosamine; 3) UTP; 4) UDP; 5) UDP-Glc; 6) UDP-Gal. Product formation was monitored by high performance anion exchange chromatography (Dionex Co., California, USA).
REFERENCES
1.Kornberg, A. (1995) J. Bacteriol.,
177, 491-496.
2.Kornberg, A., Kornberg, S. R., and Simms, E. S.
(1956) Biochim. Biophys. Acta, 20, 215-227.
3.Kornberg, S. R. (1957) Biochim. Biophys.
Acta, 26, 294-300.
4.Kyunghye, A., and Kornberg, A. (1990) J. Biol.
Chem., 265, 11734-11739.
5.Akiyama, M., Crooke, E., and Kornberg, A. (1992)
J. Biol. Chem., 267, 22556-22561.
6.Crooke, E., Akiyama, M., Rao, N. N., and Kornberg,
A. (1994) J. Biol. Chem., 289, 6290-6295.
7.Rao, N. N., and Kornberg, A. (1996) J.
Bacteriol., 178, 1394-1400.
8.Wurst, H., Shiba, T., and Kornberg, A. (1995) J.
Bacteriol., 177, 898-906.
9.Castuma, C. E., Huang, R., Kornberg, A., and
Reusch, R. N. (1995) J. Biol. Chem., 270,
12980-12983.
10.Shiba, T., Tsutsumi, K., Yano, H., Ihara, Y.,
Kameda, A., Tanaka, K., Takahashi, H., Munekata, M., Rao, N. N., and
Kornberg, A. (1997) Proc. Natl. Acad. Sci. USA, 94,
11210-11215.
11.Hengge-Aronis, R., Lange, R., Henneberg, N., and
Fischer, D. (1993) J. Bacteriol., 175, 259-265.
12.Gentry, D. R., Hernandez, V. J., Nguyen, L. H.,
Jensen, D. B., and Cashel, M. (1993) J. Bacteriol., 175,
7982-7989.
13.Cashel, M. (1994) Meth. Mol. Genet.,
3, 341-356.
14.Lange, R., Fischer, D., and Hengge-Aronis, R.
(1995) J. Bacteriol., 177, 4676-4680.
15.Tsutsumi, K., Munekata, M., and Shiba, T.,
manuscript in preparation.
16.Ruther, U., and Muller-Hill, B. (1983) EMBO
J., 2, 1791-1794.
17.Kuroda, A., Murphy, H., Cashel, M., and Kornberg,
A. (1997) J. Biol. Chem., 272, 21240-21243.
18.Rao, N. N., Liu, S., and Kornberg, A. (1998)
J. Bacteriol., 180, 2186-2193.
19.Kusano, S., and Ishihama, A. (1997) Genes
Cells, 2, 433-441.
20.Ault-Riche, D., Fraley, C. D., Tzeng, C., and
Kornberg, A. (1998) J. Bacteriol., 180, 1841-1847.
21.Blum, E., Py, B., Carpousis, A. J., and Higgins,
C. F. (1997) Mol. Microbiol., 26, 387-398.
22.Kuroda, A., and Kornberg, A. (1997) Proc.
Natl. Acad. Sci. USA, 94, 439-442.
23.Noguchi, T., and Shiba, T. (1998) Biosci.
Biotechnol. Biochem., 62, 1594-1596.
24.Bontig, C. F. C., Kortstee, J. J., and Zehnder,
A. J. B. (1991) J. Bacteriol., 173, 6484-6488.
25.Ishige, K., and Noguchi, T., submitted.
26.Noda, L. (1973) in The Enzymes (Boyer, P.
D., ed.) Academic Press, New York, Vol. 8, pp. 279-305.
27.Lu, Q., and Inoue, M. (1996) Proc. Natl. Acad.
Sci. USA, 93, 5720-5725.
28.Murata, K., Uchida, T., Kato, J., and Chibata, I.
(1988) Agric. Biol. Chem., 52, 1471-1477.
29.Wong, C.-H. (1993) U. S. Patent 551665.
30.Ito, Y., and Paulson, J. C. (1993) U. S. Patent
5409817.
31.Wong, C.-H., and Gaeta, F. C. A. (1994)
International Application Number PCT/US94/05067.
32.Miller, J. H. (1972) Experiments in Molecular
Genetics, Cold Spring Harbor Laboratory Press, Plainview, New
York.
33.Tanaka, K., Handel, K., Loewen, P. C., and
Takahashi, H. (1997) Biochim. Biophys. Acta, 1352,
161-166.
34.Manoil, C., and Kaiser, D. (1980) J.
Bacteriol., 141, 297-304.
35.Hengge-Aronis, R. (1993) Cell, 72,
165-168.
36.Walker, G. C. (1984) Microbiol. Rev.,
48, 60-93.