REVIEW: Phosphatidylinositol-3 Kinase Dependent Pathways: the Role in Control of Cell Growth, Survival, and Malignant Transformation
M. A. Krasilnikov
Institute of Carcinogenesis, Blokhin Cancer Research Center, Russian Academy of Medical Sciences, Kashirskoe Shosse 24, Moscow, 115478 Russia; fax: (095) 324-1205; E-mail: mkras@mail.cnt.ru
Received September 17, 1999
Phosphatidylinositol-3 kinase (PI3K) is one of the most important regulatory proteins that is involved in different signaling pathways and controlling of key functions of the cell. The double-enzymatic activity of PI3K (lipid kinase and protein kinase) as well as the ability of this enzyme to activate a number of signal proteins including some oncoproteins determines its fundamental significance in regulation of cell functions such as growth and survival, aging, and malignant transformation. Among the main effectors of PI3K are the mitogen-transducing signal proteins (protein kinase C, phosphoinositide-dependent kinases, small G-proteins, MAP (mitogen activated protein) kinases), which are activated either via their interaction with lipid products of PI3K or through PI3K-dependent phosphorylation of proteins. The anti-apoptotic effect of PI3K is realized by activation of proteins from another signaling pathway--protein kinase B (PKB) and/or PKB-dependent enzymes (GSK-3, ILK). PI3K plays a critical role in malignant transformation. PI3K itself possesses oncogenic activity and also forms complexes with some viral or cellular oncoproteins (src, ras, rac, alb, T-antigen), whose transforming activities are realized only in presence of PI3K. The transforming effect of PI3K is supposed to occur on the basis of complex alterations in cellular signaling pathways: appearance of constitutively generated PI3K-dependent mitogen signal and activation of some protooncogenes (src, ras, rac, etc.), PI3K/PKB-pathway stimulation resulting in delay of apoptosis and increase of cell survival, and actin cytoskeleton reorganization.
KEY WORDS: phosphatidylinositol-3 kinase, phosphoinositides, MAP kinases, protein kinase B, proliferation, apoptosis, oncogenic transformation, aging
Membrane phospholipids including phosphatidylinositol are key substances mediating the control of cell division. It was supposed for many years that phosphatidylinositol (PtdIns) and its phosphorylated derivatives (PtdIns(3)P, PtdIns(3,4)P2, and PtdIns(3,4,5)P3) took part in the transduction of mitogen signals only through their hydrolysis by phospholipase C to well known cell division mediators such as diacylglycerol and inositol phosphates. During the recent years these suggestions have been significantly changed, first of all due to the discovery and broad investigation of phosphatidylinositol-3 kinase (PI3K), the enzyme phosphorylating PtdIns in the 3-OH position of the inositol ring.
Initially PI3K was the subject of interest because of its known ability to form complexes with some viral oncoproteins such as v-src and v-ros [1, 2] and also because of involvement of intracellular PI3K in the viral transformation process [3]. Later, in 1997, the possibility of malignant transformation of cells as the result of transfection with DNA containing a fragment of a viral or cellular PI3K gene was shown [4]. Parallel investigations of biochemical properties of PI3K led to rather unexpected results. The two-subunit (regulatory p85 and catalytic p110) molecule of PI3K appears to possess both lipid kinase and protein kinase activity [5, 6]. Activation of the dimeric p85/p110 PI3K molecule occurs through phosphorylation of a tyrosine residue by either receptor (platelet, insulin-like, or epidermal growth factor receptors) or non-receptor (p60-src) tyrosine kinases [7-9]. Experiments with the use of some specific inhibitors of PI3K and/or cell transfection with different PI3K gene variants reveal PI3K being a mediator in the control of at least two very important cell functions, namely, cell division (as a necessary component of the signaling pathway initiated by growth factors) and apoptosis (the progress of which is inhibited by PI3K) [3, 10-12].
Recently, clear progress in the investigation of the mechanism of PI3K action has revealed the main mediators of its action. For instance, the event cascade leading to the delay of apoptosis is initiated by complex formation between PtdIns-phosphate products of the PI3K-catalyzed reaction and protein kinase B (PKB, also named Akt or Akt/PKB) [13, 14]. The latter enzyme plays an important role in the regulation of the activity of many genes controlling, directly or indirectly, the apoptotic process [15-17]. PI3K-mediated transduction of mitogen signal is realized in another way. In spite of the traditional view of PtdIns and its derivatives as the main components of mitogen signal, the role of PI3K in the regulation of cell division appears not to be restricted to synthesis of these compounds. Not so long ago it was shown that PI3K may directly control the activities of individual components of the RAS/RAF/ERK-mitogenic pathway by complex formation with some signal proteins; the enzyme acts in this case as a serine--threonine protein kinase [5, 6, 18].
Among the other important PI3K functions involved directly or indirectly in mitogen signal transduction, the involvement of PI3K in receptor down-regulation (endocytosis and degradation of activated growth factor receptors), in control of lysosomal enzyme synthesis [19, 20], and in reorganization of actin cytoskeleton during the course of malignant transformation process and/or mitogen stimulation of cells should also be emphasized [21].
In general, PI3K is now considered as one of the most important regulatory proteins, being involved in a number of diverse signaling pathways and controlling the main functions of the cell. PI3K activation in malignant cells after exposure to radiation or other stress [9, 22] and also the above-mentioned anti-apoptotic effect of PI3K indicate the important role of this enzyme in the control of both malignant cell resistance to damaging agents and the sensitivity of malignant tumors to chemo- or radiotherapy.
PI3K: GENERAL PROPERTIES
Phosphatidylinositol-3 kinase is a heterodimer of two subunits, catalytic and regulatory, with molecular weights of 110 kD (p110) and 85 kD (p85), respectively [9, 22]. Cloning experiments with the use of PI3K cDNA have revealed at least five isoforms of each subunit [23-27]. The regulatory p85 subunit consists of several domains including the SH3 domain, two proline rich fragments, and two SH2 domains separated by the iSH2 (inter SH2) sequence (Fig. 1). The iSH2 domain provides the interaction between the p85 and p110 subunits, and the two SH2 domains are responsible for binding of the p85/p110 heterodimer with receptor tyrosine kinases [9, 28]. It is supposed that due to the ability of the regulatory p85 subunit to interact with both the catalytic p110 subunit and receptor tyrosine kinases directed membrane targeting of p110 occurs, initiating complex formation between the enzyme and its phospholipid substrate [9, 24].
The catalytic p110 subunit of PI3K is homologous to protein kinases and possesses both serine--threonine protein kinase and phosphoinositide kinase activities [5, 6, 22, 29, 30]. Phosphorylation of PtdIns and phosphoinositides PtdIns(4)P and PtdIns(4,5)P2 occurs in the D3-position of the inositol ring leading to formation of PtdIns(3)P, PtdIns(3,4)P2, and PtdIns(3,4,5)P3, respectively. Three classes of the PI3K protein superfamily are now known. All of these possess the protein kinase activity, the difference is preferentially in the substrate specificity of the phosphoinositide kinase site. Thus, the first class includes p85/p110 heterodimers reacting with all phosphoinositides, PtdIns, PtdIns(4)P, and PtdIns(4,5)P2. These are now often referred to as phosphoinositide-3 kinases; the term reflects their substrate specificity more correctly than the traditional name, phosphatidylinositol-3 kinases. The second class involves enzymes phosphorylating preferably PtdIns and PtdIns(4)P. Finally, the third class includes PI3K that possesses additionally a specific protein transfer function and has a structural and functional resemblance with the yeast analog of PI3K, vps34p (vacuolar protein sorting). Unlike the enzymes from the first and the second classes, this one uses only PtdIns as a substrate [31-33].Fig. 1. Schematic structure of the p85/p110-heterodimer. The regulatory p85 subunit consists of (from the N-terminus) SH3-domain, two PRDs (proline rich domain), separated by BCR-homologous domain (BCR, breakpoint cluster region), and two SH2-domains, separated by the iSH2 (inter SH2)-sequence, which is responsible for binding with the p110 subunit. Arrows show the binding sites of heterodimer to the main PI3K activators: phosphotyrosine proteins (P) and small G-proteins (G).
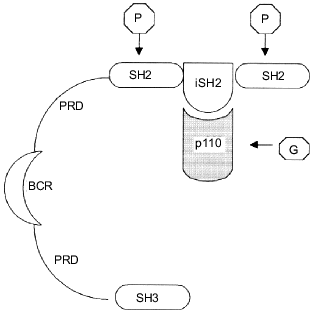
Two main processes lead to PI3K activation: p85/p110 heterodimer assembly and interaction of the heterodimer with activator proteins. As mentioned above, the binding of the catalytic and regulatory subunits occurs via the iSH2 domain of the latter. The p85/p110 heterodimer assembly does not result itself in marked enzyme activation. Moreover, some investigators reported the activity of the catalytic subunit being decreased when the p85/p110 complex is formed in vitro [28]. An additional interaction with specific activator proteins is required for the subsequent activation of the heterodimer [34-36]. The main activator proteins have tyrosine-phosphorylated amino acid sequences including both some receptor (receptors of platelet, epidermal, or insulin-like growth factors), and non-receptor (p60-src) tyrosine kinases [7-9]. The binding of phosphotyrosine sites of activator proteins with SH2 domains of the PI3K regulatory subunit causes a conformational change of the heterodimer leading to enzyme activation (Fig. 1) [28]. There are, however, other mechanisms of PI3K activation. An extra activation of the heterodimer may occur while direct interaction between the PI3K catalytic subunit and one of several cellular proteins takes place. A good example is the complex formation between p21-ras and p110 resulting in activation of PI3K [37-39]. The diversity of ways for PI3K activation to occur and also the multi-substrate specificity and double-enzymatic activity (lipid kinase and protein kinase) of the enzyme likely determine its key role in the control of cell growth and survival.
CELL GROWTH CONTROL. POSITION OF PI3K AMONG GENERAL SIGNALING
PATHWAYS
Investigations of recent years have shown the involvement of PI3K in the control of cell division being realized through at least two possible mechanisms: the first includes phosphoinositide production (PI3K lipid kinase activity), the second includes direct interaction of PI3K with some cellular signal proteins, when the protein kinase activity of PI3K may play a critical role [5, 6, 22, 29, 30].
Traditional opinions concerning the role of phosphoinositides in cell growth control were based on their role as substrates of phospholipase C, the enzyme which is activated by receptor tyrosine kinases during cell division. When accumulated in the cells due to phospholipase C action, the hydrolysis products of phosphoinositides (diacylglycerol and inositol phosphates) activate protein kinase C, thus stimulating one of the most important signaling pathways of the cell [40-43]. However, as was shown later, the phosphoinositides may have an independent significance in mitogen signal transduction, because of their ability for direct interaction with some signal proteins. The role of phosphoinositides in activation of protein kinase B (PKB), which is involved preferentially in the control of cell apoptosis, will be considered below. As for the protein mediators of cell division, protein kinase C (PKC) should be distinguished first of all. The PKC activation appears to occur not only via binding with diacylglycerol formed by hydrolysis of phospholipids, but also via the interaction with PI3K lipid products [44]. Great progress in the study of the control mechanisms of PI3K was achieved following the discovery of a new family of serine--threonine protein kinases, phosphoinositide-dependent kinases (PDK) [45, 46]. These are activated by the lipid products of PI3K, 3OH-phosphoinositides (hence the name of the family), and responsible for the phosphorylation and activation of a number of signaling protein kinases, including both PKB and PKC [45-47]. Thus, two steps of PI3K-dependent activation of protein kinase C may be distinguished: PKC interaction with diacylglycerol, the phosphoinositide hydrolysis product, and phosphorylation of PKC by PDK family enzymes (Fig. 2). Also, the data obtained from the studies on binding of phosphoinositides with SH2-containing proteins should be taken into account. These indicate one of the PI3K products, PtdIns(3,4,5)P3, interacts with SH2-domains of proteins competing with phosphotyrosine peptides [48]. The same effect may exist for an additional pathway of activation of SH2-containing signaling proteins which is independent from receptor tyrosine kinases.
The subject of particular interest is the involving of PI3K in receptor «down-stream» processes, including endocytosis and degradation of activated growth factor receptors. As known, binding of ligand with growth factor receptor and activation of its phosphotyrosine kinase domain is followed by internalization of the receptor into intracellular vesicles and its consequent degradation in lysosomes [49-51]. The whole process and, in particular, the activated receptor transfer into lysosomes appear to be under PI3K control [19, 52, 53]. For instance, the studies on the down-stream handling of platelet growth factor receptor have revealed that some mutations in the phosphotyrosine site of the receptor molecule responsible for PI3K binding may cause an almost total blockage of the receptor transfer into lysosomes [19]. The same effect is caused by wortmannin, a specific PI3K inhibitor [52, 53]. However, PI3K possesses an ability to direct control of lysosomal enzyme activity by stimulation of the transfer of de novo synthesized hydrolases into lysosomes [20].Fig. 2. Scheme displaying the involvement of PI3K in the control of intracellular signaling pathways. PI3K is activated via interaction with receptor (growth factor receptors) or non-receptor (p60-src) tyrosine kinases. Among the main "down-stream" effectors of PI3K are: PKB, responsible for anti-apoptotic signal transduction; RAS/ERK, the main mitogen-conducting pathway; RAC/JNKK/JNK, pathway partially controlling the mitogen signal transduction but involved mainly in control of other cell functions, such as stress reaction or actin cytoskeleton reorganization.
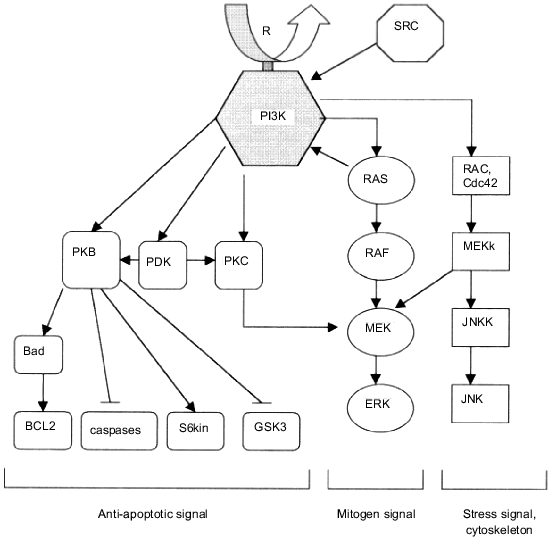
The ability of PI3K to direct binding with some cellular proteins and also the fact that the enzyme possesses not only lipid kinase, but also protein kinase activity opened new opportunities for studying its role in intracellular signaling pathways. The p85/p110 heterodimer in vivo forms complexes with a broad spectrum of cellular molecules including tyrosine kinases, Grb2, p21-ras, rac, Cdc42, tubulin, etc. [38, 39, 54-58]. The subject of principal significance for understanding the role of PI3K in the control of RAS/RAF/ERK signaling pathway is complex formation between the catalytic subunit of PI3K and p21-ras (Fig. 2). PI3K binds only with the GTP-form of ras resulting in PI3K activation observed both in vitro and in vivo [38, 39]. The same PI3K activation effect occurs when the heterodimer p85/p110 binds some other G-proteins, for instance, rac or Cdc42 [54-58]. And, on the other hand, complex formation between PI3K and p21-ras is accompanied by an increased amount of activated (GTP-bound) form of ras [59]. Moreover, the presence of PI3K appears to be necessary both for stimulation of the RAS/RAF/ERK pathway and for induced transformation of cells, and, in addition, in some cases PI3K activity inhibition may cause total blockage of transformation [21]. It should be mentioned that the mutual control between PI3K and p21-ras is rather complex and does not corresponded to a linear model of mitogen signal transfer. PI3K is supposed to activate p21-ras (possibly via membrane targeting of SOS-proteins) and is activated simultaneously via its binding with the GTP-form of ras or other G-proteins [44].
However, the significance of PI3K for the RAS/RAF/ERK signaling pathway is not limited by its influence on p21-ras. Recently, the important role of serine--threonine protein kinase activity of PI3K in the control of cellular MAP-kinases was demonstrated. Experiments using different classes of PI3K whose lipid and protein kinase activity components differ revealed that only the protein kinase activity of PI3K causes the activation of cellular MAP-kinases [18]. The level of the synthesis of phosphoinositides (lipid products of PI3K) did not influence markedly the MAP-kinase activities [18]. Thus, the general scheme of PI3K-dependent control of cellular mitogen-transduction signaling pathways consists of several stages, the main being: PI3K activation via the binding of p85/p110 heterodimer with tyrosine-phosphorylated proteins and/or small G-proteins (p21-ras, rac, Cdc42); the synthesis of 3-OH phosphoinositides which are the sources of both diacylglycerol and inositol phosphates and activators of some protein kinases (PKB, PKC, PDK) they can directly interact with; serine--threonine phosphorylation of secondary PI3K messengers and the activation of MAP-kinases (Fig. 2). Also, the scheme of PI3K involvement in cellular metabolism should be supplemented with PI3K-dependent control of stress-activated signaling pathways.
PI3K IN THE CONTROL OF APOPTOSIS
Among other cellular protein targets of PI3K, a particular role belongs to these which are involved in the cell response to stress. Moreover, although in the control of cell division PI3K plays a rather secondary role, its role in the control of cell survival and resistance to stress is a key one. This conclusion was made on the basis of the discovery and investigation of a PI3K/PKB-dependent signaling pathway [13-17, 60].
The mitogenic activity of growth factors and cytokines does not always correlate with their ability to prevent cell death. The platelet and insulin-like growth factors are good examples of compounds possessing anti-apoptotic activity. However, the fibroblast growth factor or the epidermal growth factor possessing high mitogenic activity have a negligible influence on cell survival [61-63]. A partial explanation of these facts was obtained from comparative studies on signaling pathways activated by growth factors in target cells. A stimulation of the traditional RAS/RAF/ERK-pathway usually does not result in significant anti-apoptotic effect [64, 65]. The ability to prevent apoptosis was detected in serine--threonine protein kinase B (PKB), which is activated by some growth factors [63, 64]. Studies on PKB activation pathways have shown that PI3K is a mediator of an activator signal for PKB [15-17, 64, 66].
PI3K is now considered as one of the main intracellular factors responsible for the transmission of anti-apoptotic signal and controlling the survival of cells. For instance, overexpression of PI3K in cells is accompanied by a strongly marked anti-apoptotic effect and causes a significant increase in cell survival under the influence of radiation [15-17, 65, 67]. On the contrary, PI3K specific inhibitors cause increased apoptosis and decreased cell survival [11, 12, 65, 67]. The data obtained from many experiments indicate the PKB activation by complex formation between this enzyme and lipid products of PI3K is a key event in the realization of the anti-apoptotic effect of PI3K [13-17]. There are two main stages of PKB activation: the binding of PH (pleckstrin homology)-domain of PKB with PtdIns(3)P and/or PtdIns(3,4)P2, the main products of lipid kinase reaction catalyzed by PI3K [13, 14, 66], and the phosphorylation in Thr-308 position by PDK-1 kinase (phosphoinositide-dependent kinase-1) (Fig. 2) [45, 46]. PI3K-dependent activation of PKB occurs independently from the influence of PI3K on the RAS/RAF/ERK pathway: in the first case, the binding of PKB with lipid products of PI3K is sufficient, but in the second one, as mentioned above, the involvement of the protein kinase component of PI3K in the effect of PI3K on MAP kinases is strongly necessary [18].
What is the further route of the signal from PKB and what is the nature of signaling pathways leading from PKB and controlling the survival of cells? Several mechanisms independently activated via PKB which can lead to the block of apoptosis are known at present (Fig. 2). First, it should be mentioned that no basic proteins belonging to the Bcl family of the most widely distributed negative apoptosis regulators are known among the direct targets of PKB [65]. The only exception known is Bad, which being phosphorylated by PKB is prevented from its binding to Bcl-2 [68, 69]. Proteases of the caspase family are known to be PKB mediators, which are activated during apoptosis. PKB inhibits their activities; this property may serve as a basis of its anti-apoptotic effect [65]. Another possible effector of PKB is p70 S6 kinase, which possesses a distinct anti-apoptotic property and, being phosphorylated directly by PKB kinase, displays increased activity [46, 64]. Finally, data obtained from recent investigations have revealed an important role of GSK-3 (glycogen synthetase kinase-3) in the induction of programmed cell death. The direct phosphorylation of GSK-3 by PKB kinase leading to a drastic decrease in GSK-3 activity is one of its regulatory mechanisms [70, 71].
Undoubtedly, the scheme of signal transduction pathways initiated by PI3K and PKB presented here is rather incomplete. Knowledge on the nature of secondary messengers involved in signal promotion from PI3K and PKB is expanding from day to day; new data on the role of these substances in the control of cell response to stress are appearing. For instance, new data were obtained on the involvement of integrin-associated protein kinases (ILK, integrin-linked kinase) in realization of the anti-apoptotic effect of PI3K [71]. The problem on the role of stress-activated kinases (JNK family) in the control of apoptosis and signal transduction from PI3K and PKB is under comprehensive study [72, 73].
PI3K AND CELL AGING
The ability of PI3K to control key functions of the cell such as proliferation or apoptosis became the stimulus for studying the role of PI3K in the control of cell aging, another key function of the cell. The cell aging phenomenon is known to develop after the cell achieves the Hayflick limit, i.e., after the cell has passed through a definite number of divisions to the resting stage [74]. The passage of cells to resting stage ahead of time and initiation of cell aging is appreciated by some researchers as one of the programs of defense resembling programmed cell death (apoptosis) and is activated under the influence of damaging agents [75]. The fact that some substances such as ceramide involved in mitogen signal transduction and possessing apoptotic activity may (unlike traditional mitogens) effectively influence on the rate of cell aging is indirect evidence for that assumption [76].
Now several experimental proofs have been obtained that indicate that PI3K is involved in the control of aging. First of all, experiments with Caenorhabditis elegans reveal a homology between Age1,one of the genes of aging, and the gene encoding the PI3K catalytic subunit in mammals and demonstrate the involvement of Age1 in the control of development of C. elegans [77]. Convincing evidence for the involvement of PI3K in the control of cell aging have been obtained from the experiments on normal fibroblasts in vitro. Comparative analysis of the effects of PI3K inhibitor LY2940002 and MEK-1 (kinase phosphorylating ERK1/ERK2) inhibitor PD58029 on fibroblasts has shown that both substances inhibit cell proliferation. However, only in the first case (when the PI3K inhibitor acts) cell growth retardation was accompanied by a complex of specific phenotypic alterations which are normally typical for aging fibroblasts: galactosidase activation, overexpression of collagenase gene, and decreased expression of EPC-1 gene (early population doubling level cDNA 1) which is a specific marker of proliferating fibroblasts [78]. This suggests that under PI3K-dependent control of cell aging the components of the anti-apoptotic signaling pathway controlled by PI3K and independent of the RAS/RAF/ERK cascade have the most important significance. Apparently, it is the activation of the anti-apoptotic pathway and especially PKB that mainly determines the involvement of PI3K in the control of cell aging.
PI3K IN MALIGNANT TRANSFORMATION OF CELLS
As mentioned above, initial interest in PI3K was due mostly to the involvement of PI3K in malignant transformation of cells. Some viral oncoproteins such as src, abl, T-antigen form complexes with PI3K, and the presence of PI3K in a cell is required for realization of their transforming potential. Experiments with mutant forms of those proteins have shown that the loss of their ability to form complexes with PI3K results in a dramatic decrease in the transforming activity of the oncoproteins [8, 79-82]. As a result of complex formation between oncoproteins and PI3K, the enzyme becomes activated and the level of PtdIns(3,4)P2/PtdIns(3,4,5)P3 increases; also, the transforming efficiency decreases in accordance with the decrease in intracellular concentration of phosphoinositides [3, 81, 83].
The question on the independent oncogenic activity of PI3K remained without answer for a long time, but in 1997 the viral analog of a gene encoding the PI3K catalytic subunit p110 was found in the genome of ASV 16 (avian sarcoma virus 16) [4]. Studies on the transforming potential of the gene encoding the catalytic subunit p110 named p3k have shown that both viral and cellular variants of this gene cause morphologic transformation in chicken embryo fibroblasts in vitro [4]. What components of the PI3K-induced signaling pathway take a direct part in malignant transformation of cells? Now, with no doubt, these are the lipid products of PI3K and protein kinases they activate, such as the above-mentioned protein kinase B (PKB) encoded by the Akt protooncogene. The transformation of cells by the p3k gene is shown to be accompanied by a drastic increase in both the PKB activity and the rate of phosphorylation of endogenous substrates [4]. An important role in the process of PI3K-dependent transformation belongs to the stimulation of the RAS/RAF/ERK-kinase cascade and the increase in AP-1 transcription factor activity [84]. The complex changes in cellular signaling pathways, namely, the appearance of constantly generated PI3K-dependent mitogen signal and the activation of some protooncogenes (src, ras, rac, etc.), and also PKB-dependent pathway stimulation, which leads to partial blockage of apoptosis and increased survival of cells, are possibly the basis for the transforming effect of PI3K (Fig. 2).
Studies on the role of PI3K in the formation of the actin cytoskeleton deserve separate attention. It is well known that both the induction of cell proliferation and the malignant transformation of cells are accompanied by reorganization of the actin cytoskeleton [21, 85-87]. Data from recent studies indicate that one of the key elements in the control of this process is PI3K. It has been shown that PI3K activation appears to be an essential condition for reorganization of actin filaments in a cell. Oncoprotein rac, which belongs to the small G-protein group and is involved in signal transduction via the stress-activated kinases of JNK family, is considered as a possible effector of PI3K (Fig. 2) [54-58, 88-90]. The other possible pathway for PI3K to control actin assembly is based on the lipid kinase activity of PI3K. The main role in this pathway belongs to one of the PI3K substrates, PtdIns(4,5)P2, which interacts with actin binding proteins, thus stimulating the actin polymerization process [55, 87, 91-94]. Intracellular activation of PtdIns(4,5)P2 metabolizing enzymes (phospholipase C and PI3K), in particular, under the influence of mitogenic/oncogenic factors, leads to a decreased amount of PtdIns(4,5)P2 bound with actin and, as a result, to actin depolymerization [95, 96].
The discovered oncogenic activity of PI3K is evidence for an important role of the enzyme in carcinogenesis and tumor growth. In fact, some data indicate changes in phosphoinositide level during malignant tumor progression [97]. Our study on PI3K expression in breast cancer tumors has revealed in 79% of cases a significant activation of PI3K compared to adjacent normal tissue [98]. It was recently shown that the effect of some tumor growth suppressor genes are activated via PI3K inhibition. One of these is a product of suppressor gene product PTEN/MMCA1, a phosphatase which dephosphorylates PtdIns(3,4,5)P3 [99]. Specific inhibitors of PI3K (wortmannin, LY 294002) cause a significant retardation of cell growth in culture and prevent the cells from being malignantly transformed in vitro [84, 100, 101]. However, the problem concerning the anti-tumor activity of PI3K antagonists or inhibitors and, on the whole, their usage in cancer therapy is far from completely resolved. Further investigations are required.
In general, the progress achieved in recent years in studying PI3K allows us to place this enzyme among the main signaling proteins of the cell. The diversity of ways for PI3K activation and also its unique biochemical properties (multisubstrate specificity and both lipid and protein kinase activity) determine its critical role in the control of key functions in the cell: growth and survival, aging, malignant transformation. The main PI3K effectors are mitogen-transducing signaling proteins (protein kinase C, phosphoinositide-dependent kinases, small G-proteins, MAP-kinases) which are activated either via interaction with lipid products of PI3K or through PI3K-dependent phosphorylation of proteins [38, 39, 45-47, 54, 58]. The anti-apoptotic effect of PI3K is realized through the activation of proteins from another regulatory pathway, the protein kinase B (PKB) and PKB-dependent enzymes (GSK-3, ILK) (Fig. 2) [13-17, 70, 71].
Nevertheless, the mechanism of some PI3K effects, such as PI3K-dependent control of malignant transformation, remain unclear. PI3K possesses a direct oncogenic activity and also potentiates the effects of other oncogenes activating and increasing the transforming activity of many of the known oncoproteins (ras, rac, Akt, src) [8, 79-83]. A number of cell damaging factors cause a drastic increase in PI3K activity [9, 15-17, 22]. That activation of PI3K leads to delay of apoptosis in cells with damaged DNA structure and also causes an extra activation of cellular oncoproteins and stimulates uncontrolled cell growth and, hence, may have perhaps great significance in carcinogenesis.
The problem of malignant transformation is closely related to another PI3K function, the control of cell aging. A decrease in PI3K activity causes an increase in aging rate in normal cells [78], but the mechanism of PI3K-dependent control of cell aging and the role of individual effectors of PI3K are still unknown. It may be that, as in the case of PI3K-induced malignant transformation, the activation of mitogen-dependent proteins and (as appears to be the most important) the anti-apoptotic signal constantly generated by PI3K play an important role in the control of aging. However, studies in this field are just beginning, and it is unclear whether the PI3K activity changes during cell aging and the overexpression of PI3K leads to significant delay in the aging process. Undoubtedly, in the near future studies will answer these and other questions on the mechanism of the effect of PI3K in normal and malignant cells.
It should be noted in conclusion that PI3K, when being identified as one of the key signaling proteins, lets us tie together many events which occur in cells under the action of mitogenic or oncogenic factors and also stress, and, at first glance, seem to be independent. Every year new data appear concerning the description of novel PI3K effectors or revealing a correlation between known cell proteins and PI3K-dependent signaling pathways. We are on the way to understanding the basic principles of coordinated control of biochemical signaling pathways and their significance for fundamental cell properties: growth, aging, transformation.
This study was supported by the Russian Foundation for Basic Research (grant No. 98-04-48200).
REFERENCES
1.Sugimoto, Y., Whitman, M., Cantley, L. C., and
Erikson, R. L. (1984) Proc. Natl. Acad. Sci. USA, 81,
2117-2121.
2.Macara, I. G., Marinetti, G. V., and Balduzzi, P.
C. (1984) Proc. Natl. Acad. Sci. USA, 81, 2728-2732.
3.Cantley, L. C., Auger, K., Carpenter, C. L.,
Duckworth, B., Graziani, A., Kapeller, R., and Soltoff, S. (1991)
Cell, 64, 281-302.
4.Chang, H. W., Aoki, M., Fruman, D., Auger, K.,
Bellacosa, A., Tsichlis, P., Cantley, L. C., Roberts, T., and Vogt, P.
(1997) Science, 276, 1848-1850.
5.Carpenter, C. L., Auger, K., Duckworth, B., Hou,
W., Schaffhausen, B., and Cantley, L. C. (1993) Mol. Cell.
Biol., 13, 1657-1665.
6.Dhang, R., Hiles, I., Panayotou, G., Roche, S.,
Fry, M., Gout, I., Totty, N., Truong, O., Vicendo, P., Yonesawa, K.,
Kasuga, M., Courtneidge, S., and Waterfield, M. (1994) EMBO J.,
13, 522-533.
7.Ruderman, N., Kapeller, R., White, M., and Cantley,
L. (1990) Proc. Natl. Acad. Sci. USA, 87, 1411-1415.
8.Fukui, Y., and Hanafusa, H. (1989) Mol. Cell.
Biol., 9, 1651-1658.
9.Kapeller, R., and Cantley, L. C. (1994)
BioEssays, 16, 565-576.
10.Yao, R., and Cooper, G. M. (1995) Science,
267, 2003-2006.
11.Scheid, M. P., Lauener, R. W., and Duronio, V.
(1995) Biochem. J., 312, 159-162.
12.Minshall, C., Arkins, S., Freund, G. G., and
Kelley, K. W. (1996) J. Immunol., 156, 939-947.
13.Franke, T. F., Kaplan, D. R., Cantley, L. C., and
Toker, A. (1997) Science, 275, 665-668.
14.Klippel, A., Kavanaugh, W., Pot, D., and
Williams, L. T. (1997) Mol. Cell Biol., 17, 338-345.
15.Kauffmann-Zeh, A., Rodrigues-Vicciana, P.,
Urlich, E., Gilbert, G., Coffer, P., Downward, J., and Evan, G. (1997)
Nature, 385, 544-548.
16.Khwaja, A., Rodrigues-Vicciana, P., Wennstrom,
S., Warne, P., and Downward, J. (1997) EMBO J., 16,
2783-2793.
17.Kulik, G., Klippel, A., and Weber, M. (1997)
Mol. Cell. Biol., 17, 1595-1606.
18.Bondeva, T., Pirola, L., Burgarelli-Leva, G.,
Rubio, I., Wetzker, R., and Wymann, M. (1998) Science,
282, 293-296.
19.Joly, M., Kazlauskas, A., Fay, F., and Corvera,
S. (1994) Science, 263, 684-687.
20.Brown, W., DeWald, D., Emr, S., Plutner, H., and
Balch, W. (1995) J. Cell. Biol., 130, 781-796.
21.Rodrigues-Vicciana, P., Warne, P., Khwaja, A.,
Marte, B., Pappin, D., Das, P., Waterfield, M., Ridley, A., and
Downward, J. (1997) Cell, 89, 457-467.
22.Carpenter, C. L., Duckworth, B., Auger, K.,
Cohen, B., Schaffhausen, B., and Cantley, L. C. (1990) J. Biol.
Chem., 265, 19704-19711.
23.Otsu, M., Hiles, L., Gout, I., Fry, M.,
Ruiz-Larrea, F., Panayotou, G., Thompson, A., Dhang, R., Hsuan, J.,
Totty, N., Courtneidge, S., and Waterfield, M. (1991) Cell,
65, 91-104.
24.Skolnik, E., Margolis, B., Mohammadi, M.,
Lowenstein, E., Fisher, R., Drepps, A., Ulrich, A., and Schlessinger,
J. (1991) Cell, 65, 83-90.
25.Escobedo, J., Navakasattusas, S., Kavanaugh, W.,
Milfay, D., Freid, V., and Williams, L. (1991) Cell, 65,
75-82.
26.Pons, S., Asano, T., Glasheen, E., Miralpeix, M.,
Zhang, Y., Fisher, T., Myers, M., Sun, X., and White, M. (1995) Mol.
Cell. Biol., 15, 4453-4465.
27.Inukai, K., Anai, M., Van Breda, E., Hosaks, T.,
Katagiri, H., Funaki, M., Fukushima, Y., Ogihara, T., Yazaki, Y.,
Kikuchi, M., Oka, Y., and Asano, T. (1996) J. Biol. Chem.,
271, 5317-5320.
28.Yu, J., Wjasow, C., and Backer, J. (1998) J.
Biol. Chem., 273, 30199-30203.
29.Whitman, M., Downes, C., Keeler, M., and Cantley,
L. (1988) Nature, 332, 644-646.
30.Auger, K., Serunian, L., Soltoff, S., Libby, P.,
and Cantley, L. (1989) Cell, 57, 167-175.
31.Stoyanov, B., Volinia, S., Hanck, T., Rubio, I.,
Loubtchenkov, M., Malek, D., Stoyanova, S., Vanhaesebroeck, B., Dhang,
R., and Nurnberg, B. (1995) Science, 269, 690-693.
32.MacDougall, L., Domin, J., and Waterfield, M.
(1995) Curr. Biol., 5, 1404-1415.
33.Schu, P., Takegawa, K., Fry, M., Stack, J.,
Waterfield, M., and Emr, S. (1993) Science, 260,
88-91.
34.Backer, J., Myers, J., Shoelson, S., Chin, D.,
Sun, X., Miralpeix, M., Hu, P., Margolis, B., Skolnik, E., Shlessinger,
J., and White, M. (1992) EMBO J., 11, 3469-3479.
35.Carpenter, C., Auger, K., Chanudhuri, M., Yoakim,
M., Schaffhausen, B., Shoelson, S., and Cantley, L. (1993) J. Biol.
Chem., 268, 9478-9483.
36.Rordorf-Nikolic, T., Van Horn, D., Chen, D.,
White, M., and Backer, J. (1995) J. Biol. Chem., 270,
3662-3666.
37.Kodaki, T., Woscholski, R., Hallberg, B.,
Rodriguez-Viciana, P., Downward, J., and Parker, P. (1994) Curr.
Biol., 4, 798-806.
38.Rodriguez-Viciana, P., Warme, P., Dhang, R.,
Vanhaesebroeck, B., Gout, I., Fry, M., Waterfield, M., and Downward, J.
(1994) Nature, 370, 527-532.
39.Rodriguez-Viciana, P., Warme, P., Vanhaesebroeck,
B., Waterfield, M., and Downward, J. (1996) EMBO J., 15,
2442-2451.
40.Orr, W., and Newton, J. (1994) J. Biol.
Chem., 269, 27715-27723.
41.Cazaubon, F., Bornancin, F., and Parker, P.
(1994) Biochem. J., 301, 443-451.
42.Hug, H., and Sarre, T. (1993) Biochem. J.,
291, 329-334.
43.Kopnin, B. P. (2000) Biochemistry
(Moscow), 65, 2-27.
44.Carpenter, L., and Cantley, L. (1996) Biochim.
Biophys. Acta, 1288, 11-16.
45.Alessi, D., Deak, M., Casamayor, A., Caudwell,
F., Morrice, N., Norman, D., and Gaffney, P. (1997) Curr. Biol.,
7, 776-789.
46.Alessi, D., Kozlowski, M., Weng, Q.-P., Morrice,
N., and Auruch, J. (1998) Curr. Biol., 8, 69-81.
47.Le Good, J., Ziegler, W., Parekh, D., Alessi, D.,
Cohen, P., and Parker, P. (1998) Science, 281,
2042-2045.
48.Rameh, L., Chen, C., and Cantley, L. (1995)
Cell, 83, 821-830.
49.Chang, C., Lazar, C., Walsh, B., Komuro, M.,
Kuhn, L., and Tainer, J. (1993) J. Biol. Chem., 268,
19312-19320.
50.Lamaze, C., and Schmid, S. (1995) J. Cell.
Biol., 129, 47-54.
51.Opresko, L., Chang, C., Will, B., Burke, P.,
Gill, G., and Wiley, H. (1995) J. Biol. Chem., 270,
4325-4333.
52.Joly, M., Kazlauskas, A., and Corvera, S. (1995)
J. Biol. Chem., 270, 13225-13230.
53.Shpetner, H., Joly, M., Hartley, D., and Corvera,
S. (1996) J. Cell. Biol., 132, 595-605.
54.Zheng, Y., Bagrodia, S., and Cerione, R. (1994)
J. Biol. Chem., 269, 18727-18730.
55.Tolias, K., Cantley, L., and Carpenter, C. (1995)
J. Biol. Chem., 270, 17656-17659.
56.Wennstorm, S., Siegbahn, A., Yokote, K.,
Arvidsson, A., Heldin, C., Mori, S., and Claesson, W. (1994)
Oncogene, 9, 651-660.
57.Nobes, C., Hawkins, P., Stephens, L., and Hall,
A. (1995) J. Cell. Sci., 108, 225-233.
58.Hawkins, P., Eguinova, A., Qiu, R., Stokoe, D.,
Cooke, F., Walters, R., Wennstrom, S., Claesson-Welsh, L., Evans, T.,
Symons, M., and Stephens, L. (1995) Curr. Biol., 5,
393-403.
59.Hu, P., Margolis, B., Skolnik, E., Lammers, R.,
Ullrich, A., and Schlessinger, J. (1992) Mol. Cell. Biol.,
12, 981-990.
60.Gerber, H., McMurtrey, A., Kowalski, J., Yan, M.,
Keyt, B., Dixit, V., and Ferrara, N. (1998) J. Biol. Chem.,
273, 30336-30343.
61.Fairbairn, L., Cowling, G., Reipert, B., and
Dexter, T. (1993) Cell, 74, 823-832.
62.Raff, M., Barres, B., Burne, J., Coles, H.,
Ishizaki, Y., and Jacobson, M. (1993) Science, 262,
695-700.
63.Harrington, E., Bennett, M., Fanidi, A., and
Evan, G. (1994) EMBO J., 13, 3286-3295.
64.Burgering, B., and Coffer, P. (1995)
Nature, 376, 599-602.
65.Kennedy, S., Wagner, A., Conzen, S., Jordan, J.,
Bellacosa, A., Tsichlis, P., and Hay, N. (1977) Genes Dev.,
11, 701-713.
66.Franke, T., Yang, S., Chan, T., Datta, K.,
Kazlauskas, A., Morrison, D., Kaplan, D., and Tsichlis, P. (1995)
Cell, 81, 727-736.
67.Krasilnikov, M., Adler, V., Fuchs, S., Dong, Z.,
Haimovitz-Friedman, A., Herlyn, M., and Ronai, Z. (1999) Mol.
Carcinog., 24, 64-69.
68.Zha, J., Harada, H., Yang, E., Jockel, J., and
Kosmeyer, S. (1996) Cell, 87, 619-628.
69.Zundel, W., and Giaccia, A. (1998) Genes
Dev., 12, 1941-1946.
70.Cross, D., Aless, D., Cohen, P., Andjelkovich,
M., and Hemmings, B. (1995) Nature, 378, 785-789.
71.Delcommenne, M., Tan, C., Gray, V., Rue, L.,
Woodgett, J., and Dedhar, S. (1998) Proc. Natl. Acad. Sci. USA,
95, 11211-11216.
72.Kharbanda, S., Saleem, A., Shafman, T., Emoto,
Y., Taneja, N., Rubin, E., Weichselbaum, R., Woodgett, J., Avruch, J.,
Kyriakis, J., and Kufe, D. (1995) J. Biol. Chem., 270,
18871-18874.
73.Lopez-Ilasaca, M., Li, W., Uren, A., Yu, J.,
Kazlauskas, A., Gutkind, J., and Heidaran, M. (1997) Biochem.
Biophys. Res. Commun., 232, 273-277.
74.Hayflick, L., and Moorhead, P. (1961) Exp.
Cell Res., 25, 585-621.
75.Wenberg, R. (1997) Cell, 88,
573-575.
76.Venable, M., Lee, J., Smyth, M., Bielawska, A.,
and Obeid, L. (1995) J. Biol. Chem., 270,
30701-30708.
77.Morris, J., Tissenbaum, H., and Ruvkin, G. (1996)
Nature, 382, 536-539.
78.Tresini, M., Mawal-Dewan, M., Cristofalo, V., and
Sell, C. (1998) Cancer Res., 58, 1-4.
79.Whitman, M., Kaplan, R., Schaffhausen, B.,
Cantley, L., and Roberts, T. (1985) Nature, 315,
239-242.
80.Courtneidge, S., and Heber, A. (1987)
Cell, 50, 1031-1043.
81.Varticovski, L., Daley, G., Jackson, P.,
Baltimore, D., and Cantley, L. (1991) Mol. Cell. Biol.,
11, 1107-1113.
82.Freund, R., Dawe, C., Carrol, J., and Benjamin,
T. (1992) Am. J. Pathol., 141, 1409-1417.
83.Fukui, Y., Saltiel, A., and Hanafusa, H. (1991)
Oncogene, 6, 407-411.
84.Huang, C., Schmid, P., Ma, W., Schmid, H., and
Dong, Z. (1997) J. Biol. Chem., 272, 4187-4194.
85.Rovensky, Yu. A. (1998) Biochemistry
(Moscow), 63, 1029-1043.
86.Bar Sagi, D., and Feramisco, J. (1986)
Science, 233, 1061-1068.
87.Janmey, P. (1994) Ann. Rev. Physiol.,
56, 169-191.
88.Minden, A., Lin, A., Claret, F., Abo, A., and
Karin, M. (1995) Cell, 81, 1147-1157.
89.Hill, C., Wynne, J., and Treisman, R. (1995)
Cell, 81, 1159-1170.
90.Coso, O., Chiariello, M., Yu, J., Teramoto, H.,
Crespo, P., Xu, N., Miki, T., and Gutkind, J. (1995) Cell,
81, 1137-1146.
91.Hartwig, J., Bokoch, G., Carpenter, C., Janmey,
P., Taylor, L., Toker, A., and Stossel, T. (1995) Cell,
82, 643-653.
92.Tolias, K., Couvillon, A., Cantley, L., and
Carpenter, C. (1997) Mol. Cell. Biol., 18, 762-770.
93.Chong, L., Traynor, K., Bokoch, G., and Schwartz,
M. (1994) Cell, 79, 507-513.
94.Shibasaki, Y., Ishihara, H., Kizuki, N., Asano,
T., Oka, Y., and Yazaki, Y. (1997) J. Biol. Chem., 272,
7578-7581.
95.Fukami, K., Furuhashi, K., Inagaki, M., Endo, T.,
Hatano, S., and Takenawa, S. (1992) Nature, 359,
150-152.
96.Fukami, K., Endo, T., Imamura, M., and Takenawa,
T. (1994) J. Biol. Chem., 269, 1518-1522.
97.Bordoni, A., Hrelia, S., Biagi, P., and Berra, B.
(1992) Int. J. Cancer, 50, 402-404.
98.Gershtein, E., Shatskaya, V., and Krasilnikov, M.
(1999) Clin. Chim. Acta, 287, 59-67.
99.Wu, X., Senechal, K., Neshat, M., Whang, Y., and
Sawyers, C. (1998) Proc. Natl. Acad. Sci. USA, 95,
15587-15591.
100.Valhos, C., Matter, W., Hui, K., and Brown, R.
(1994) J. Biol. Chem., 269, 5241-5248.
101.Yano, H., Nakanishi, S., Kimura, K., Hanai,
Saitoh, Y., Fukui, Y., Nonomura, Y., and Matsuda, Y. (1993) J. Biol.
Chem., 268, 25846-25856.