Phenoptosis: Programmed Death of an Organism
V. P. Skulachev
Department of Bioenergetics, Belozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (7-095) 939-0338; E-mail: skulach@head.genebee.msu.su
Received September 3, 1999
Programmed cell death (apoptosis) is well-established in many multicellular organisms. Apoptosis purifies a tissue from cells that became useless or even harmful for the organism. Similar phenomena are already described also at subcellular level (suicide of mitochondria, i.e., mitoptosis) as well as at supracellular level (degradation of some organs temporarily appearing in the course of ontogenesis and then disappearing by means of apoptosis of the organ-composing cells). Following the same logic, one may put a question about programmed death of an organism as a mechanism of purification of a kin, community of organisms, or population from individuals who became unwanted for this kin, etc. A putative mechanism of such kind is proposed to be coined “phenoptosis” by analogy with apoptosis and mitoptosis. In a unicellular organism (the bacterium Escherichia coli), three different biochemical mechanisms of programmed death are identified. All of them are actuated by the appearance of phages inside the bacterial cell. This may be regarded as a precedent of phenoptosis which prevents expansion of the phage infection among E. coli cells. Purification of a population from infected individuals looks like an evolutionary invention useful for a species. Such an invention has high chances to be also employed by multicellular organisms. Most probably, septic shock in animals and humans serves as an analog of the phage-induced bacterial phenoptosis. It is hypothesized that the stress-induced ischemic diseases of brain and heart as well as carcinogenesis if they are induced by repeated stresses also represent phenoptoses that, in contrast to sepsis, are age-dependent. There are interrelations of programmed death phenomena at various levels of complexity of the living systems. Thus, extensive mitoptosis in a cell leads to apoptotic death of this cell and extensive apoptosis in an organ of vital importance results in phenoptotic death of an individual. In line with this logic, some cases are already described when inhibition of apoptosis strongly improves the postischemic state of the organism.
KEY WORDS: programmed cell death, apoptosis, mitochondria, septic shock, stroke, heart attack, carcinogenesis, programmed death of an organism
Abbreviations: PTP) permeability transition pore; ROS) reactive oxygen species; TNF) tumor necrosis factor; zDEVD-fmk) N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethyl ketone; zVAD-fmk) N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone; zYVAD-fmk) N-benzyloxycarbonyl-Tyr-Val-Ala-Asp-fluoromethyl ketone.
During the last few years, the phenomenon of programmed cell death
(apoptosis) became one of the most popular topics of biological
studies. Apoptosis proved to be involved in numerous mechanisms of
defense against pathogens and toxic agents as well as in ontogenesis,
immune response, etc. (for reviews, see [1-4]).
Analogous phenomena have been described at the subcellular level. Disintegration of reticulocyte mitochondria, initiated by activation of mitochondrial 15-lipoxygenase, an event accompanying maturation of reticulocytes to erythrocytes [5], can be an example of programmed death of mitochondria defined (by analogy with apoptosis) as “mitoptosis” [4, 6]. It was suggested that the ROS-induced opening of the permeability transition pores (PTP) in the inner mitochondrial membrane represents a mitoptotic mechanism [7] purifying the mitochondrial population in the cell from those mitochondria that produce large amounts of ROS [1, 8].
For sure, apoptosis is a way to purify cellular populations in tissues from unwanted or useless cells. It is not surprising, therefore, that ROS-induced mitoptosis, if it becomes a large-scale process, results in apoptosis purifying a tissue from ROS-overproducing cells [1, 4, 6].
A large-scale apoptosis in a tissue or an organ results in elimination of this tissue (organ), as it takes place in tadpole or human embryo at a stage of ontogenesis when the tail disappears1.
1 For instance, Kashiwagi et al. [66] have recently shown that addition of thyroxin (a hormone which in tadpoles is known to cause regression of the tail) results in shortening of severed tails incubated in special medium. The following chain of events was elucidated: thyroxine --> NO synthase induction --> [NO]^ --> inactivation of catalase (and, probably, glutathione peroxidase) by NO --> [H2O2]^ --> apoptosis.
Following the same logic, one may put a question concerning the possibility of programmed cell death at the level of the organism. Programmed death of organ(s) of vital importance, if it exists, must lead to the death of the organism. In this paper some observations will be summarized suggesting the existence of programmed death of organisms which I have named, by analogy with apoptosis and mitoptosis, “phenoptosis” [9].
PHENOPTOTIC PRECEDENT IN BACTERIA
It is obvious that the aims of apoptosis (purification of an organism from unwanted cells) can be achieved only if the organism in question is multicellular. Therefore a programmed cell death of a unicellular organism must be regarded as phenoptosis.
Martin Raff wrote in his recent mini-review on apoptosis: "The best understood example of non-animal cell suicide occurs in bacteria. Some strains of Escherichia coli produce an inactive form of a protease. If they become infected by a particular virus, one of the viral polypeptides binds to the protease and activates it. The activated protease cleaves and inactivates a bacterial protein required for protein synthesis (EF-Tu), shutting down synthesis and killing the bacterium--thereby curtailing viral multiplication and protecting the nearby E. coli [population] from infection [10-12]. Although this bacterial death program shares a number of features with caspase-dependent apoptosis in animal cells, the proteases involved in the two programmed processes are unrelated" [3].
It has been found that E. coli possesses also at least two more mechanisms of the same function. One of them includes the identical events in the beginning of the suicide cascade, but the final product is the tRNALys-specific ribonuclease instead of the EF-Tu-specific protease [13]. The third suicide mechanism was found to produce the RexA and RexB proteins where the former is a phage DNA receptor, the latter being an ion channel activated by the DNA·RexA complex. The channel is assumed to dissipate all the ion gradients of the bacterial membrane and, as a result, kills the cell [14, 15]. Interestingly, all these systems are encoded by “parasitic” elements of the E. coli DNA including prophages and plasmids (for review, see [12]).
Purification of the population is of apparent biological advantage. Therefore, it is hardly specific for E. coli. Rather, as a useful invention, it was retained in the course of evolution also in higher organisms.
SEPTIC SHOCK: PHENOPTOSIS IN HIGHER ORGANISMS?
A higher organism strongly infected by a dangerous pathogen is an unwanted guest for a kin, community, or population of these organisms just as a phage-infected cell of E. coli is for an E. coli population. Quick death of the strongly infected individual might be a cruel but radical solution of the problem from the population point of view. Perhaps, septic shock is a suicide mechanism (phenoptosis) purifying an animal population or a human society from dangerously infected individuals.
All the characteristics of septic shock indicate that the death of an ill organism is well organized by the organism itself, while the role of the pathogen is rather passive. So-called endotoxin causing sepsis is a lipopolysaccharide forming the wall of a Gram-negative bacterium. Toxicity of endotoxin is absolutely dependent on the endotoxin-binding protein in blood and some receptors in the plasma membrane of the human or animal cells. Sepsis is accompanied by massive formation of tumor necrosis factor (TNF) and other cytokines inducing apoptosis. Knock-out of genes coding for these protein factors or inhibition of receptors decreases the endotoxin efficiency (for reviews, see [16, 17]). It seems quite probable that septic shock will be impossible when we identify and block all the receptors involved in processing of the phenoptotic signal caused by lipopolysaccharide. As for bacterial lipopolysaccharide per se, it seems to be not toxic at all. It is not dangerous until it can be recognized by the macroorganism, being a signal of appearance in blood and tissues of Gram-negative bacteria. Such bacteria are especially dangerous because they are protected by their lipopolysaccharide wall against attack by antibacterial systems of the organism.
Certainly, phenoptosis is the last line of the defense of the population. If the level of the pathogen in an individual is not so high as to become a serious danger for the population, the same signal (bacterial lipopolysaccharide) is used to attract phagocytes to the infected region of the organism. Separate lines of antibacterial defense are the lipopolysaccharide-induced formation of the above-mentioned cytokines as well as uncoupling proteins responsible for hyperthermia up to body temperatures non-permissible for some pathogenic bacteria. If the infection degree is not too high, all these measures are certainly useful for the organism. This is why the common opinion concerning sepsis consists in that it represents an over-use by the macroorganism of its defensive antimicrobial tools [16, 17]. Such an explanation is, in fact, identical to that commonly employed to account for over-use of apoptosis in lethal cases of stroke or heart attack (see below). However, there is a difference, important in this context, between sepsis, on one hand, and stroke and heart attack, on the other. Sepsis, in fact, does not depend so much as stroke and heart attack upon the age of the patient.
Within the framework of this point of view, it remains, however, quite unclear why a control mechanism preventing such a potentially very dangerous system from killing the organism, was not invented in the course of biological evolution. On the other hand, the phenoptosis concept easily explains the septic shock in the same fashion as the already described programmed death of the phage-infected E. coli cells (see above). Apparently, the principal difference of the two systems consists in that in E. coli we deal with mechanisms absolutely specialized in phenoptosis, whereas human phenoptosis, i.e., large-scale cell suicide, develops from a limited apoptosis which is initially useful for the organism.
THE MYSTERY OF REPERFUSION
The main mitochondrial process, oxidative phosphorylation, as well as the majority of other functions of these organelles, become impossible when O2 is exhausted. It is not surprising therefore that injuries after anoxia were initially attributed to the depletion of ATP due to the arrest of oxidative phosphorylation and inhibition of other mitochondrion-dependent processes. Such a point of view proved to be right for long-term anoxia. However, injury appearing after short-term anoxia seems to be related to the subsequent reoxygenation rather than to the preceding anaerobiosis. The crucial role in the reoxygenation-induced injury is performed by ROS [18-20]. It is an effect of this kind that is most probably responsible for the death of the brain after several minutes of ischemia. During such a short period of time, no serious changes in levels of ATP and other key metabolites are detected.
To account for such a dramatic phenomenon, as well as for unfavorable action of reoxygenation in general, I would like to put forward the following suggestion.
It seems probable that the ROS-linked injury is mainly due to effects of hydroxyl radical which is very much more aggressive and has very much more positive redox potential (+1.35 V) than, say, O2-. or H2O2. The major source of OH. in the cell is the so-called the Fenton reaction [21]:
The intracellular concentration of free iron ions is very small, i.e., less than 5·10-6 M [22]. Under aerobiosis, most of them are in the oxidized form (Fe3+) since Fe2+ is autoxidized in the presence of O2, the process being strongly accelerated by the formation of complexes of Fe2+ and citrate and some other carboxylates always present in the cell. This is why the rate of the Fenton reaction in vivo is usually very low even if H2O2 is available.
The situation dramatically changes under anaerobic conditions. Here Fe2+ autoxidation becomes impossible by definition (O2 is absent) and Fe3+ is immediately reduced to Fe2+ by various metabolites always having more negative redox potential than the Fe2+/Fe3+ pair (+0.77 V). Such a process should require a very short time because of the small intracellular concentration of iron ions. The appearance of Fe2+ is not dangerous until O2 is unavailable. However, reoxygenation actuates the Fenton reaction and OH. is formed.
Again, the damage caused by the OH. formed in such a way should be limited in time by the small amount of free iron ions. Nevertheless, such an event as a burst of OH. production is apparently recognized by the cell even if it is short. There are some reasons to assume that the cell always monitors the level of such most aggressive compound as hydroxyl radical. This might, e.g., be done by measuring of the oxidized phosphatidylserine in the outer leaflet of the plasma membrane by a special phosphatidylserine receptor similar to that found in phagocytes [4].
A cell producing OH. not only severely injures itself but becomes very dangerous for the tissue and, therefore, it and its closest neighbors must be eliminated as soon as possible. Thus, a burst of the OH. formation at reoxygenation should initiate an apoptotic program in both the OH.-producing cell and bystanders. Massive death of cells in the reoxygenated organ will be a direct consequence of realization of this program.
It has been shown that reperfusion of, e.g., liver after ischemia results in a burst of ROS formation entailed with apoptosis, necrosis, and subsequent inflammation [23-25]. Mitochondria are somehow involved in this effect since recombinant adenoviral expression of mitochondrial Mn-dependent superoxide dismutase in mouse liver prior to lobal ischemia--reperfusion was found to significantly lower subsequent acute liver damage [26].
A burst of ROS formation was observed by Sluse and coworkers in isolated mitochondria during postanoxic reoxygenation [27]. Under the same conditions, a cyclosporin A-sensitive PTP opening was described by Tanaka et al. [28]. According to Leducq et al. [29], liver reperfusion causes further decrease in the ATP level which was already lowered in ischemia. A cyclosporin A addition to perfusate results in an ATP level increase on reperfusion. A great cyclosporin A-induced increase in the viability of hepatocytes after reperfusion was described by Lemasters and his colleagues [30].
It has been reported by many laboratories that iron chelators strongly increase the resistance of cells and organs to oxidative stress (for references, see [20, 31]). This is in line with the above described hypothesis explaining the dramatic effect of reperfusion by the Fe2+-catalyzed OH. formation.
Another reason for the reperfusion effect may be linked to an anaerobiosis-induced increase in the xanthine oxidase level. Both the activity and the quantity of this enzyme in cytosol is reversibly proportional to [O2] (for references, see [4]). When oxygen comes to an anaerobic cell, it meets with much higher xanthine oxidase than under aerobic conditions. Aerobiosis will decrease the xanthine oxidase level but this needs some time. As a result of such relationships, the rate of xanthine oxidase-catalyzed O2-. and H2O2 production is strongly increased during the first minutes of reoxygenation [20]. In combination with the increased Fe2+/Fe3+ ratio, elevated [O2-.] and [H2O2] can entail a really dramatic enhancement of the OH. formation. Therefore, it is not surprising that an inhibitor of xanthine oxidase, allopurinol, possesses pronounced therapeutic effect at reperfusion [20, 32-34].
It should be taken into account that the increase in the ROS level under reperfusion causes some secondary effects that make the situation ever worse. In particular, the appearance of oxidized phosphatidylserine on the surface of the ROS-producing cell attracts phagocytes that start to bombard this cell with O2-. generated by NADPH oxidase in their plasma membrane. This results in further increase in [ROS] in the target cell and in all the region surrounding such a cell. Moreover, ROS are known to initiate the formation and release of proinflammatory agents, such as cytokines, that subsequently attract and activate neutrophils (polymorphonuclear leukocytes) carrying out phagocytosis [20].
BRAIN AND HEART ISCHEMIA
Recent studies on brain and heart ischemia clearly demonstrate that the delayed impairment phase after both stroke and heart attack is at least partially a consequence of the reoxygenation-induced apoptosis. Extensive apoptosis is described after ischemia followed by reperfusion in the immature [35-37] and adult brain (for review, see [38]). It was also revealed as a result of reperfusion in heart muscle [39-42].
From the point of view of the organism, two alternative possibilities should be taken into account.
1. The reoxygenation-induced apoptosis is useful since it eliminates dangerous cells producing ROS.
2. It is harmful since so many cells are killed that normal functioning of an organ of vital importance becomes impossible.
Paradoxically, it is the second alternative that proved to be valid. To discriminate between the two above cases, very simple logic was employed. In the first case inhibitors of apoptosis must make the state of the patient worse whereas in second case the same treatment must improve the situation.
During last two years, several reports were published indicating that arrest of apoptosis by specific caspase inhibitors is of clear favorable effect helping to survive after reoxygenation. These most important findings are summarized in the next section.
ARREST OF APOPTOSIS HELPS TO OVERCOME THE CONSEQUENCES OF
REOXYGENATION
Endres et al. [43] studied the consequences of 10 min cerebral artery occlusion and subsequent reperfusion on mouse brain. It was found that intracerebroventricularly injected caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD-fmk), as well as the other caspase inhibitor N-benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethyl ketone (zDEVD-fmk) decreased infarct size, neurological deficits, and the number of apoptotic cells in brain when administrated 6 h after reperfusion. Both the infarct volume and DNA cleavage (an event accompanying apoptosis) were reduced by the caspase inhibitors by factors of 2-3. Injection of the inhibitors 18 h after reperfusion proved to be ineffective (see also [44]).
zVAD-fmk is known to inhibit a wide spectrum of caspases whereas zDEVD-fmk specifically suppresses the activity of caspase 3. N-Benzyloxycarbonyl-Tyr-Val-Ala-Asp-fluoromethyl ketone (zYVAD-fmk), another peptidyl protease inhibitor which does not affect caspase 3, failed to prevent the brain ischemia consequences in rat as was recently reported by Chen et al. [45]. In this study, effects of cerebral ischemia--reperfusion on hippocampal neurone survival, apoptosis, and caspase 3 level were investigated. It was found that intraventricular infusion of zDEVD-fmk, but not of zYVAD-fmk, before or 2 h after ischemia resulted in a strong decrease of all the ischemic effects measured. For instance, the zDEVD-fmk pretreatment caused almost 10-fold enhancement of the neurone survival and 4-fold decrease in the number of apoptotic cells. In the pretreated rats, caspase 3 was activated by ischemia by factor 2 instead of 10 in the control.
Caspase inhibitors proved to be also effective in traumatic brain injury [46].
Yaoita et al. [47] described favorable action of zVAD-fmk at heart reperfusion. Rats were subjected to a 30 min coronary occlusion followed by a 24 h reperfusion. zVAD-fmk was administrated intravenously every 6 h starting at 30 min before the coronary occlusion until 24 h of reperfusion. It was found that zVAD-fmk strongly improves hemodynamic characteristics of the heart impaired after reperfusion. The infarct size/ischemic area decreased from 67 to 53% and the number of apoptotic cardiomyocytes in the ischemic area was reduced from 11 to 3%.
Summarizing the present state-of-the-art, Thornberry and Lazebnik [48] concluded that “peptidyl caspase inhibitors are effective in animal models of stroke, myocardial ischemia--reperfusion injury, liver disease, and traumatic brain injury”.
It was already mentioned that zDEVD-fmk inhibits caspase 3, which operates at the final steps of the apoptotic cascade. There are some indications that similar protective effects can be obtained by inhibiting the apoptotic cascade in the very beginning, i.e., at stage of the plasma membrane receptors (for reviews, see [38, 48, 49]).
Moreover, the protection can also be organized at the intermediate level of, e.g., proapoptotic protein kinase JNK. To activate apoptosis, JNK should be phosphorylated by another kinase (SEK-2). There is a protein phosphatase dephosphorylating JNK-P. Cytosolic heat shock protein Hsp70, according to Sherman and his colleagues [50], activates the JNK-P phosphatase and prevents the JNK-P-dependent apoptosis. This effect was found to be different from the Hsp70 chaperon action. Apparently the above mechanism allows the cell to monitor the level of denatured proteins to initiate apoptosis when their concentration increases so strongly that all the Hsp70 molecules appear to be bound to denatured proteins rather than to JNK-P phosphatase [50].
It was found that transgenic mice expressing the human Hsp70 have improved post-ischemic myocardial recovery [51, 52]. It seems possible that the same effect is involved in the favorable action of myocardial adaptation to ischemia induced by repeated cyclic short-term ischemic episodes (so-called preconditioning). On the other hand, it was shown that expression of the anti-apoptotic proteins, NF-kappaB and Bcl-2, is induced by the above adaptation [53, 54]. One of the anti-apoptotic actions of Bcl-2 is found to consist of inhibition of the release of cytochrome c from mitochondria [55].
Another way to prevent reoxygenation-induced apoptosis is to decrease the ROS level by treatment with antioxidants.
The concept of the role of apoptosis in ischemia--reperfusion injury is summarized in the figure.
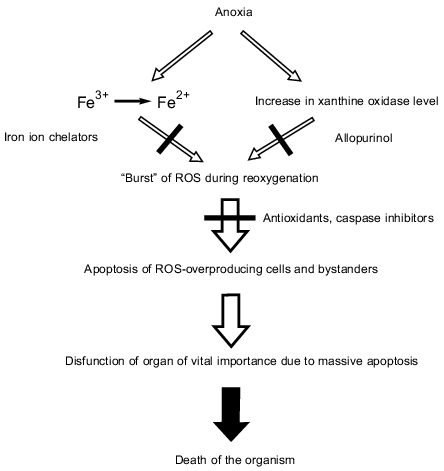
Dramatic consequences of anoxia mediated by ROS and apoptosis. Even short-term ischemia resulting in anoxia (anaerobiosis) is sufficient (1) to reduce free iron ions from Fe3+ to Fe2+ and (2) to increase the xanthine oxidase level. When O2appears (reoxygenation at reperfusion), xanthine oxidase produces O2-. and H2O2 that are converted to OH. mediated by Fe2+ oxidation. Besides xanthine oxidase, mitochondria and other ROS-producing systems contribute to the O2-. and H2O2 formation (not shown in the figure). Effects of Fe2+ and xanthine oxidase can be abolished by chelators and allopurinol, respectively. A burst of ROS including its most aggressive form (OH.) results in apoptosis of the ROS-overproducing cells and bystanders that are attacked by the ROS overproducer-formed H2O2. Massive apoptosis results in dysfunction of an organ of vital importance (e.g., heart or brain) causing death of the organism.
PROGRAMMED AGE-DEPENDENT DEATH OF THE HUMAN ORGANISM; WEISMANN'S
AND LEWIS' HYPOTHESES
More than a century ago, August Weismann hypothesized that death of old individuals had been invented by biological evolution as a kind of adaptation. He wrote: “Worn-out individuals are not only valueless to the species, but they are even harmful, for they take the place of those which are sound...
I consider that death is not a primary necessity, but that it has been secondarily acquired as an adaptation. I believe that life is endowed with a fixed duration, not because it is contrary to its nature to be unlimited, but because the unlimited existence of individuals would be a luxury without any corresponding advantage” [56].
Weismann's idea was strongly criticized by Medawar [57] who mainly analyzed possible reasons for death of wild animals (according to Medawar, the life span of animals living under natural conditions is too short to allow the age-dependent constituent of mortality to have a measurable contribution). However, the possible adaptive origin of aging has never been forgotten by gerontologists as an alternative to the generally accepted point of view on age-dependent death as an inevitable consequence of a break in such a complex system as the living organism (for review, see [58]).
It seems obvious that for long-lived animals like primates, any increase in the percentage of old individuals in a population is undesirable not only for the reason indicated by Weismann but also due to a risk of deterioration of the population's genofond by the genomes of elders containing mutations accumulated during the long period of life.
Discovery of programmed death of living cells gave a new impetus to the development of Weismann's concept. In 1997 I reconsidered his hypothesis within the framework of the newest observations concerning apoptosis [9]. Later Kim Lewis [59] published a paper postulating a concept which includes the programmed, age-dependent death of human individuals. He assumed that “transmission of knowledge from grandparents to progeny served as a driving force for extending human longevity”, explaining in this way why maximal duration of life for humans is two-times longer than for higher monkeys. He has also postulated that in old humans a special mechanism of programmed death can operate which is switched on when an old individual appears to be useless for others. Lewis writes: “In humans, older individuals in early societies who are no longer useful could increase reproductive success by activating this programmed aging mechanism, which would result in channelling resources to progeny. Decreased emotional support and mastery are mortality risk factors in the elderly, supporting this hypothesis of programmed death in humans”. Really, according to Penninx et al. [60], there is a correlation between mortality and such psychological factors as lack of emotional support and the understanding that the person no longer controls his or her life [59].
It seems probable that factors other than those of psychological nature may also be reasons for the age-dependent phenoptosis in humans. I think so since apparently the programmed death of the organism is also inherent at least in some plants and animals. For instance, it is well known that a bamboo lives for 15-20 years using vegetative multiplication, then comes into bloom and dies when the seeds ripen. The male of some squids species dies just after transferring his spermatophores to a female [61] (for discussion, see [9]).
STROKE, HEART ATTACK, AND CANCER AS MECHANISMS OF AGE-DEPENDENT
PHENOPTOSIS
As an example of a molecular mechanism responsible for programmed age-dependent human death, Lewis [59] considered Hsp70-controlled apoptosis. He mentioned an observation of Udelsman et al. [62] who showed that Hsp70 was induced in rat aortic smooth muscle cells in response to restraining stress, and this response diminished in old animals. It seems probable that in old animals all the Hsp70 pool, which is not already induced by stress, will be bound to denatured proteins whose level elevates under the stress conditions. As a result, the amount of Hsp70 bound to the JNK-P phosphatase (see above) decreases, the phosphatase activity lowers, [JNK-P] rises and, hence, apoptosis is actuated. Such an effect can be of catastrophic consequences if even a rather small ischemic area arise in the brain or heart, entailing massive apoptotic death by the mechanism shown in the figure. In such a context, stroke and heart attack in old people may be considered as age-dependent phenoptosis rather than an over-use of the apoptosis-linked antioxidant defense mechanism (K. Lewis, personal communication).
It is remarkable that JNK is involved in one of the TNF-induced apoptotic pathways. The dramatic role of TNF in both stroke and heart attack is well known. Thus, TNF and other cytokines seem to be competent in inducing: 1) mitoptosis (via PTP-linked burst of ROS production in mitochondria); 2) apoptosis (by means of PTP-dependent mitochondrial swelling and release of pro-apoptotic factors from mitochondria to cytosol), and 3) phenoptosis (due to large-scale apoptosis mediated by both JNK and mitochondria in septic shock and brain and heart muscle ischemia).
Disappearance with age of the stress-linked induction of Hsp-70 is hardly the only phenoptotic mechanism. Stress per se is always dangerous since it causes a rise of the ROS level due to disbalance in the ROS-generating and ROS-scavenging processes. This stimulates both ROS-induced damages to mitochondrial DNA (and other ROS-sensitive cellular components) and apoptosis [63].
According to Kapahi et al. [64], the life span of various mammals positively correlates with the in vitro resistance of their fibroblasts to oxidative stress and some other stresses. Thus, the probability of phenoptosis inevitably increases in old peoples subjected to stresses because of the above-mentioned psychological factors or of some simpler reasons such as, e.g., inability to perform work which could easily be done previously.
It seems possible that cancer, the third reason for death of old people (after brain and heart ischemia) can also play the role of a phenoptotic mechanism. It might be operative in those individuals whose anti-apoptotic system is still dominating over the pro-apoptotic system even in old age. It is generally assumed that apoptosis may be one of the lines of the anticancer defense. A steady decrease in apoptosis means an increased risk of carcinogenesis. It is also known that stresses in some way stimulate carcinogenesis, and cancer, like ischemia, is a disease of old rather than of young people [65]. This does not mean that stroke, heart attack, or cancer can occur in old age only. Within the framework of the above concept, not only the age but also absolute number of stresses during the life and the level of the anti-stress defense mechanisms are of importance. Moreover, it is obvious that, e.g., cancer can appear in an age-independent fashion as a result of uptake of cancerogenic xenobiotics. Lethal cases can also be consequences of a damage of phenoptosis-controlling mechanisms.
It is noteworthy that all of the above-listed diseases, if not treated in time in a proper way, are fast and lethal, differing in this respect from numerous chronic diseases slowly developing because of accumulation of metabolic and structural defects in old humans.
To summarize the concept described above, one may assume that septic shock is an age-independent phenoptosis which purifies the population of badly infected individuals whereas stroke, heart attack, and cancer in old people may be mechanisms of the stress-linked, age-dependent phenoptosis eliminating old individuals that are not only useless but are harmful for the genofond. All the listed cases can be classified as (1) hyperapoptotic when apoptosis is enhanced to a level inconsistent with the life of the organism (sepsis, ischemic brain and heart diseases) or (2) hypoapoptotic when malignant cells are no longer eliminated by apoptosis (cancer).
POSSIBLE STRATEGY OF ANTIPHENOPTOTIC DEFENCE
The above consideration represents further development of an idea [63] that evolution of life resulted in formation of specialized biochemical mechanisms the operation of which results in a situation when interests of an individual is sacrificed for the sake of a group of individuals or entire population. Since Homo sapiens does not rely upon the rate of evolution, which is too slow for him, mechanisms of this kind look now as a harmful atavism. Elimination of such an atavism might help to overcome some human diseases and among them those that are related to phenoptosis.
Within the framework of this hypothesis, a novel approach to the solution of some basic problems of modern medicine can be formulated. The plan of actions might consist of the following.
1. Identification of cases of phenoptosis in humans (Septic shock? Age-dependent stroke, heart attack, cancer?...).
2. Elucidation of basic causes inducing phenoptosis. Here special attention should be paid to analysis of psychological factors that initiate in an organism the biochemical suicide program.1
1 It is symbolic that causes of phenoptosis mentioned by Lewis [59], namely, lack of emotional support and loss of personal independence are considered, if we deal with old people, among main motifs of suicide in the literal sense of this awful word.
3. Elimination, if possible, of causes of phenoptosis.
4. Description of physiological and biochemical mechanisms responsible for realization of phenoptosis. All the mechanisms of actuating and carrying out apoptosis in organs of vital importance should be elucidated.
5. For hyperapoptotic phenoptoses: abrogation of the apoptotic signal and cessation of already started apoptotic program by specific inhibitors. When trying to prolong human life in such cases, we should try to break a limited number of programmed death pathways rather than to repair numerous injuries in such a very complicated system as the human organism. This approach, I hope, has a chance to be promising at least in certain cases since, as we say here in Russia, “it is easier to break than to repair”.
I am very grateful to Professor Kim Lewis for very fruitful discussion and for sending me a preprint of his paper. I also thank Dr. Boris Chernyak and Dr. Michael Sherman for useful advice.
Addendum. After submission of this paper, an article by Migliaccio et al. appeared (Nature (1999), 402, 309-313) showing that treatments of mouse fibroblasts by ROS causes serine phosphorylation in p66shc protein and apoptosis. In fibroblasts from mice without a gene coding for p66shc, ROS failed to induce apoptosis. These mice live 30% longer eating the same amount of food as the control animals. Such a phenomenon can well be accounted for by the scheme: ROS --> phospho-p66shc --> apoptosis --> phenoptosis.
REFERENCES
1.Skulachev, V. P. (1996) Quart. Rev.
Biophys., 29, 169-202.
2.Jacobson, M. D., Weil, M., and Raff, M. C. (1997)
Cell, 88, 347-354.
3.Raff, M. C. (1998) Nature, 396,
119-122.
4.Skulachev, V. P. (1999) Mol. Asp. Med.,
20, 139-184.
5.Schnurr, K., Hellwing, M., Seidemann, B., Jungblut,
P., Kühn, H., Rapoport, S. M., and Schewe, T. (1996) Free
Radic. Biol. Med., 20, 11-21.
6.Skulachev, V. P. (1998) Biochim. Biophys.
Acta, 1363, 100-124.
7.Zorov, D. B., Kinnally, K. W., and Tedesci, H.
(1992) J. Bioenerg. Biomembr., 24, 119-124.
8.Skulachev, V. P. (1994) Biochemistry
(Moscow), 59, 1433-1434.
9.Skulachev, V. P. (1997) Biochemistry
(Moscow), 62, 1191-1195.
10.Yu, Y.-T. N., and Snyder, L. (1994) Proc.
Natl. Acad. Sci. USA, 91, 802-806.
11.Bergsland, K. J., Kao, C., Yu, Y.-T. N., Gulati,
R., and Snyder, L. (1990) J. Mol. Biol., 213,
477-494.
12.Snyder, L. (1995) Mol. Microbiol.,
15, 415-420.
13.David, M., Borasio, G. D., and Kaufmann, G.
(1982) Proc. Natl. Acad. Sci. USA, 79, 7097-7101.
14.Matz, K., Schmandt, M., and Gussin, G. N. (1982)
Genetics, 102, 319-327.
15.Parma, D. H., Snyder, M., Sobolevsky, S., Brody,
E., and Gold, L. (1992) Genes Devel., 6, 497-510.
16.Klosterhalfen, B., and Bhardaj, R. S. (1998)
Gen. Pharm., 31, 25-32.
17.Fenton, M. J., and Gelonbock, D. T. (1998) J.
Leukocyte Biol., 64, 25-32.
18.McCord, J. M. (1985) N. Engl. J. Med.,
312, 159-163.
19.Bulkley, G. B. (1987) Brit. J. Cancer,
8, 66-73.
20.Hochman, A. (1998) in Oxidative Stress in
Skeletal Muscle (Reznick, A. Z., et al., eds.) Birkhauser Verlag,
Basel, pp. 239-256.
21.Hippeli, S., and Elstner, E. F. (1999) FEBS
Lett., 443, 1-7.
22.Flitter, W., Rowley, D. A., and Halliwell, B.
(1983) FEBS Lett., 158, 310-312.
23.Arthur, M. J., Bentley, I. S., Tanner, A. R.,
Saunders, P. K., Millward-Sadler, G. H., and Wright, R. (1985)
Gastroenterology, 89, 1114-1122.
24.Atalla, S. L., Toledo-Pereyra, L. H., MacKenzie,
G. H., and Cederna, J. P. (1985) Transplantation, 40,
584-590.
25.Mathews, W. R., Guido, D. M., Fischer, M. A., and
Jaeschke, H. (1994) Free Radic. Biol. Med., 16,
763-770.
26.Zwacka, R. M., Zhou, W., Zhang, Y., Darby, Ch.
J., Dubus, L., Halldorson, J., Oberley, L., and Engelhardt, J. F.
(1998) Nature Med., 4, 698-704.
27.Du, G., Mouithys-Mickalad, A., and Sluse, F. E.
(1998) Free Radic. Biol. Med., 25, 1066-1074.
28.Tanaka, T., Hakoda, S., and Takeyama, N. (1998)
Free Rad. Biol. Med., 25, 26-32.
29.Leducq, N., Delmas-Beauvieux, M.-V.,
Bourdel-Marchasson, I., Dufour, S., Gallis, J.-L., Canioni, P., and
Diolez, P. (1998) Biochem. J., 336, 501-506.
30.Qian, T., Nieminen, A.-L., Herman, B., and
Lemasters, J. J. (1997) Am. J. Physiol., 273,
C1783-C1792.
31.Breuer, W., Epsztejn, S., and Cabantchik, Z. I.
(1996) FEBS Lett., 382, 304-308.
32.Smith, J. K., Carden, D. L., and Korthuis, R. J.
(1989) Am. J. Physiol., 257, H1782-H1789.
33.McCutchan, H. J., Schwappach, J. R., Enquist, E.
G., Walden, D. L., Terada, L. S., Reiss, O. K., Leff, J. A., and
Repine, J. E. (1990) Am. J. Physiol., 258,
H1415-H1419.
34.Concannon, M. J., Dooley, T. W., and Puckett, C.
L. (1991) Microsurgery, 12, 18-22.
35.Ferrer, I., Tortosa, A., Macaya, A., Sierra, A.,
Moreno, D., Munell, F., Blanco, R., and Squier, W. (1994) Brain
Pathol., 4, 115-122.
36.Mehmet, H., Yue, X., Squier, M. V., Lorek, A.,
Cady, E., Penrice, J., Sarraf, C., Wylezinska, M., Kirkbride, V.,
Cooper, C., Brown, G. C., Wyatt, J. S., Reynolds, E. O. R., and
Edwards, A. D. (1994) Neurosci. Lett., 181, 121-125.
37.Beilharz, E. J., Bassett, N. S., Sirimanne, E.
S., Williams, C. E., and Gluckman, P. D. (1995) Brain Res. Mol.
Brain Res., 29, 81-91.
38.Taylor, D. L., Edwards, A. D., and Mehmet, H.
(1999) Brain Pathol., 9, 93-117.
39.Gottlieb, R. A., Burleson, K. O., Kloner, R. A.,
Babior, B. M., and Engler, R. L. (1994) J. Clin. Invest.,
94, 1621-1628.
40.Fliss, H., and Gattinger, D. (1996) Circ.
Res., 79, 949-956.
41.Maulik, N., Kagan, V. E., Tyurin, V. A., and Das,
D. K. (1998) Am. J. Physiol., 274, H272-H278.
42.Maulik, N., Yoshida, T., and Das, D. K. (1998)
Free Rad. Biol. Med., 24, 869-875.
43.Endres, M., Namura, S., Shimizu-Sasamata, M.,
Waeber, C., Zhang, L., Gomez-Isla, T., Hyman, B. T., and Moskowitz, M.
A. (1998) J. Cerebral Blood Flow Metab., 18, 238-247.
44.Hara, H., Friedlander, R. M., Gagliardini, V.,
Ayata, C., Fink, K., Huang, Z., Shimizu-Sasamata, M., Yuan, J., and
Moskowitz, M. A. (1997) Proc. Natl. Acad. Sci. USA, 94,
2007-2012.
45.Chen, J., Nagayama, T., Jin, K., Stetler, R. A.,
Zhu, R. L., Graham, S. H., and Simon, R. P. (1998) J. Neurosci.,
18, 4914-4928.
46.Yakovlev, A. G., Knoblach, S. M., Fan, L., Fox,
G. B., Goodnight, R., and Faden, A. I. (1997) J. Neurosci.,
17, 7415-7424.
47.Yaoita, H., Ogawa, K., Maehara, K., and Maruyama,
Y. (1998) Circulation, 97, 276-281.
48.Thornberry, N. A., and Lazebnik, Y. (1998)
Science, 281, 1312-1316.
49.Barigana, M. (1998) Science, 281,
1302-1303.
50.Gabai, V. L., Meriin, A. B., Yaglom, J. A.,
Volloch, V. Z., and Sherman, M. Yu. (1998) FEBS Lett.,
438, 1-4.
51.Plumier, J. C., Ross, B. M., Currie, R. W.,
Angelidis, C. E., Kazlaris, H., Kollias, G., and Pagoulatos, G. N.
(1995) J. Clin. Invest., 95, 1854-1860.
52.Radford, N. B., Fina, M., Benjamin, I. J.,
Moreadith, R. W., Graves, K. H., Zhao, P., Gavva, S., Wiethoff, A.,
Sherry, A. D., Malloy, C. R., and Williams, R. S. (1996) Proc. Natl.
Acad. Sci. USA, 93, 2339-2342.
53.Maulik, N., Yoshida, T., Engelman, R. M., Deaton,
D., Flack, J. E., 3rd, Rousou, J. A., and Das, D. K. (1998) Mol.
Cell Biochem., 186, 139-145.
54.Maulik, N., Goswami, S., Galang, N., and Das, D.
K. (1999) FEBS Lett., 443, 331-336.
55.Skulachev, V. P. (1998) FEBS Lett.,
423, 275-280.
56.Weismann, A. (1889) Essays upon Heredity and
Kindred Biological Problems, Claderon Press, Oxford.
57.Medawar, P. B. (1952) An Unsolved Problem of
Biology, H.K. Lewis, London.
58.Kirkwood, T. B., and Cremer, T. (1982) Hum.
Genet., 60, 101-121.
59.Lewis, K. (1999) Mech. Ageing Dev.,
109, 43-51.
60.Penninx, B. W., van Tilburg, T., Kriegsman, D.
M., Deeg, D. J., Boeke, A. J., and van Eijk, J. T. (1997) Am. J.
Epidem., 146, 510-519.
61.Nesis, K. N. (1997) Cruel Love Among the
Squids. In Russian Science: Withstand and Revive
(Byalko, A. V., ed.) [in Russian], Nauka-Fizmatlit, Moscow.
62.Udelsman, R., Li, D. G., Stagg, C. A., and
Holbrook, N. J. (1995) J. Gerontol. Ser. A, Biol. Sci. Med.
Sci., 50, B187-B192.
63.Skulachev, V. P. (1998) Biochemistry
(Moscow), 63, 1335-1343.
64.Kapahi, P., Boulton, M. E., and Kirkwood, T. B.
L. (1999) Free Rad. Biol. Med., 26, 495-500.
65.Anisimov, V. N., and Solov'ev, M. V. (1999)
The Evolution of Concepts in Gerontology [in Russian], Eskulap,
St. Petersburg.
66.Kashiwagi, A., Hanada, H., Yabuki, M., Kanno, T.,
Ishisaka, R., Sasaki, J., Inoue, M., and Utsumi, K. (1999) Free Rad.
Biol. Med., 26, 1001-1009.