Structure and Folding of Bacteriophage T4 Gene Product 9 Triggering Infection. I. Production and Properties of Recombinant Protein
G. A. Navruzbekov1, L. P. Kurochkina1*, V. A. Kostyuchenko1, T. G. Zurabishvili2, S. Y. Venyaminov3, and V. V. Mesyanzhinov1
1Shemyakin and Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, ul. Miklukho-Maklaya 16/10, Moscow, 117871 Russia; fax: (095) 336-6022; E-mail: vvm@ibch.siobc.ras.ru2Ivanovskii Institute of Virology, Russian Academy of Medical Sciences, ul. Gamalei 16, Moscow, 123098 Russia
3Department of Biochemistry and Molecular Biology, Mayo Foundation, 200 First Street SW, Rochester, Minnesota, 55905, USA
* To whom correspondence should be addressed.
Received July 15, 1999
Gene product 9 (gp9, 288 amino acid residues per monomer, molecular weight 30.7 kD) of bacteriophage T4 triggers the baseplate reorganization and the sheath contraction after interaction of the long tail fibers with the receptors of the bacterial cell. In this work we have produced the recombinant protein and determined that gp9 is a stable homotrimer and active in in vitro complementation assay completing the defective phage particles which lack gp9. According to CD-spectroscopy data, the gp9 polypeptide chain contains 65-73% beta-structure and 11-16% alpha-helical segments, this being in good agreement with secondary structure prediction results. Additionally, we have constructed a set of plasmid vectors for expression of gp9 deletion mutants. The fragments with consecutive truncations of the N-terminus of the molecule, as well as the full-length protein, are trimers resistant to SDS treatment and decrease infective phage particle formation in in vitro complementation assay with native gp9. The deletion of the molecule C-terminal region results in failure of trimerization and decreases the stability of the protein.
KEY WORDS: bacteriophage T4, gene product 9, expression vector
Abbreviations: CD) circular dichroism; IPTG) isopropyl beta-D-thiogalactoside; HAP) hydroxyapatite.
Bacteriophage T4 is a convenient model for studying the mechanisms of
molecular complex assembly. In our work we study the baseplate of T4,
which consists of several different proteins, coded by late genes [1]. Native baseplate has a hexagonal shape and is
composed of six identical wedges (arms), each being a complex of seven
oligomeric proteins, and of the central hub consisting of eight
proteins including a structural lysozyme coded by gene 5. Gp9 is the
key protein that triggers the baseplate structural reorganization and
presumably consolidates wedges, binds to the baseplate in the final
stages of the phage assembly forming the long tail fiber (LTF)
attachment sites.
The adsorption of a phage on an E. coli cell begins with the reversible interaction of LTFs and the lipopolysaccharide receptors on the bacterial surface. When at least three of the LTFs are attached to their receptors, a signal is transmitted through gp9 to the baseplate and to the short tail fibers (STF). The STFs, which are usually folded under the baseplate, extend and irreversibly bind to the corresponding receptors, resulting in global structural rearrangement of the baseplate from a hexagon shape state to a six-pointed “star”. This in turn initiates sheath contraction with subsequent injection of genomic DNA into the bacterial cell [2]. This suggests that gp9 transfers a signal from the LTFs to the baseplate.
The stability of T4 particles lacking gp9 (9--particles) and LTFs is lower than that of the wild-type phages [3]; they spontaneously contract their sheaths. However, for the particles which contain gp9 and lack LTFs, the baseplate remains unchanged in the hexagonal shape. Therefore, gp9 serves both as a signal transducer and a blocker of spontaneous rearrangement of the baseplate in the absence of the LTFs [3].
The purpose of this work was to study the structure, topology, folding, and functional details of gp9. We have constructed a plasmid vector for expression of recombinant gp9 and purified it to apparent homogeneity. Recombinant gp9 is active in in vitro complementation assay completing defective 9--particles. Based on SDS-PAGE data, gp9 is a stable trimer resistant to SDS treatment. A high content of beta-structure was proposed for gp9 by CD-spectroscopy data as well as secondary structure prediction. To determine the roles of different parts of the protein, a set of gp9 deletion mutants was constructed. Based on the resistance of produced proteins to SDS treatment and their behavior in in vitro complementation assays of 9--particles with native gp9, we conclude that the C-terminal part of the protein is important for formation of a biologically active protein trimer.
MATERIALS AND METHODS
Bacterial strains. The E. coli Top10 strain was used for the plasmid DNA preparation and transformation. The genes cloned into the plasmids were expressed in the E. coli BL21(DE3) strain under the control of the bacteriophage T7 promoter [4]. The E. coli CR63 strain was used as the permissive host, and E. coli Be/1 was used as the restrictive host.
Cloning of gene 9 and its fragments. Full-length DNA of gene 9 and its fragments with consecutive truncations were produced by PCR using oligonucleotides with point mutations to generate suitable cloning sites. The deletion fragments coding the mutants NDelta20, NDelta54, NDelta167 which lack N-terminal 20, 54, and 167 amino acid residues, respectively, and CDelta113 without 113 C-terminal amino acid residues were constructed. Gene 9 fragment coding protein NDelta167 was cloned into the NdeI--BamHI sites of pET-22b(+) vector; all the other fragments including the full-size gene 9 were cloned into the NcoI--BamHI sites of pET-23d(+) vector (Novagene, USA).
Polymerase chain reaction. A multi-channel amplifier (DNK MS2, Russia) was used to run the PCR. The reaction mixture contained (in 100 µl): ~1 ng of the wild-type T4 DNA template, 50 pmoles of each primer, 10 µl 10x PCR buffer (Promega, USA), 200 µmoles of each dNTP, 2-5 units of Taq-polymerase (Promega). Mineral oil (50 µl) was overlaid on the mixture. In all cases the reactions were started from preliminary DNA denaturation (94°C, 3 min); then 30 amplification cycles were run (94°C, 1 min; 52°C, 1 min; 72°C, 1 min). The reactions were completed at 72°C for 5 min. The amplified products were analyzed by electrophoresis in 1% agarose gel.
Expression of gp9 and its fragments in E. coli BL21(DE3). The protein gp9 was expressed as described in [4]. Competent BL21(DE3) cells were transformed by the plasmids, plated on L-agar containing 1% glucose and ampicillin (100 µg/ml), and incubated at 37°C for 12-18 h. The cells were inoculated in 2x TY medium containing ampicillin (200 µg/ml) and grown at 37°C until A600 ~ 1. Isopropyl beta-D-thiogalactoside (IPTG) was added for induction of the expression to a final concentration of 1 mM and the culture was incubated additionally for 3-4 h. The cells were centrifuged at 4000 rpm for 15 min (Megafuge 2.0 R, Heraeus Instruments, Germany).
Electrophoretic analysis of the recombinant proteins. A 100 µl sample of cell culture after expression was mixed with 30 µl of 4x sample buffer (0.25 M Tris-HCl, pH 6.8, 8% SDS, 40% (v/v) glycerol, 20% (v/v) 2-mercaptoethanol, 0.02% (w/v) bromphenol blue), heated for 3 min at 100°C, centrifuged, and 10-30 µl were loaded on a gel. SDS electrophoresis was run in 15% polyacrylamide gel according to Laemmli's method [5]. Gels were stained with 0.3% Coomassie brilliant blue R-250 (Sigma, USA) in acetic acid--ethanol--water (1:3:6 v/v) solution and destained by 50% ethanol and 7% acetic acid solution. As a marker, the standard protein mixture LMW (Pharmacia, Sweden), containing alpha-lactalbumin (14.4 kD), trypsin inhibitor (20.1 kD), carboanhydrase (30.0 kD), ovalbumin (43.0 kD), albumin (67.0 kD), and phosphorylase B (94 kD), was used.
Preparation of the cell lysates. A 0.5-ml sample of cell culture was pelleted, resuspended in 100 µl of DMM (10-fold diluted M9 salt buffer containing 20 mM MgSO4), and incubated with lysozyme (0.5 mg/ml) and DNase I (0.05 mg/ml) for 10 min at room temperature. The mixture then was subjected to three cycles of freezing in liquid nitrogen with subsequent thawing on a water bath at 30°C. The lysate was centrifuged at 12,000g for 2 min and the supernatant was used in in vitro complementation assay.
Preparation of concentrated suspensions of 9--particles. A phage plaque from top agar was extracted and transferred into 0.5 ml of physiological solution containing 20 mM MgSO4 and incubated for at least 1 h at room temperature; this suspension was used for infecting 5 ml of liquid culture of E. coli CR63 in an exponential growth phase with 2·108 cells per ml with following incubation for 16 h at 25°C. The cells were lysed by adding 2-3 drops of chloroform. The debris was removed by centrifuging at 15,000 rpm for 10 min. The titer of infective particles was (4-5)·1010 plaque forming units per ml.
Preparation of defective extracts. Cell extracts containing 9--particles of T4 phage were prepared as described in [6]. The E. coli Be/1 cells were grown in 50 ml of 2x TY medium containing 5 mM MgSO4 at 37°C to a density of 2**·108 cells/ml and infected by the amber mutant C215 with multiplicity of 5. After incubation for 15 min at 30°C the concentration of MgSO4 was increased to 20 mM and the culture was additionally incubated for 20 min. The cells were pelleted at 4000 rpm for 20 min and resuspended in 2 ml of DMM containing 0.05 mg/ml DNase I. Aliquots were stored at -70°C.
Complementation assay in vitro. A 20-µl sample of defective extract was mixed with a 20-µl sample of cell lysate containing the expressed protein. The mixture was incubated for 2 h at 30°C. During incubation 5 µl aliquots were 1000-fold diluted in physiological solution and titrated using E. coli CR63 cells. To study the influence of gp9 deletion mutants on the formation of infective particles, the cell lysates were incubated with the mutants for 30 min at 30°C, followed by the addition of the full-size protein.
Purification of the recombinant proteins. The cells from 200-500 ml of culture were resuspended in 4-10 ml of 20 mM Tris-HCl buffer, pH 7.8, and incubated for 20 min at room temperature with lysozyme (0.5 mg/ml). The cell suspension was sonicated for 2 min (in cycles of 15 sec pulses followed by 15 sec pauses) using an UD-20 ultrasonic disintegrator (Techpan, Poland). The cell debris was removed by centrifuging for 10 min at 12,000g. Nucleic acids and accompanying proteins were removed by adding 10% (w/v) polyethylenimine and 4 M NaCl to final concentrations of 0.1% and 0.2 M, respectively. The mixture was incubated for 1 h (4°C). The precipitate was removed by centrifuging under the same conditions. Saturated (NH4)2SO4 was added gradually to the supernatant to the final concentration of 30-45% for protein precipitation, followed by incubation for 1 h at 4°C. The protein precipitate was collected by centrifugation, dissolved in 10 mM Tris-HCl buffer, pH 7.8, and dialyzed against the same buffer overnight. The solution was applied to a column (1 × 5 cm) with hydroxyapatite (HAP) (Bio-Rad, USA) equilibrated with 10 mM sodium phosphate buffer, pH 7.8. The protein was eluted by the same buffer and the fractions were analyzed by electrophoresis.
UV spectroscopy. Absorption spectra were recorded at 25°C on a Cary 219 (Varian, USA) spectrophotometer in 240-400 nm wavelength range at the resolution of 1 nm. All spectra were corrected for turbidity [7]. The absorption coefficient was obtained by the nitrogen micromethod [8], assuming the nitrogen content of 16.6%. The measurements were recorded in 0.1 M sodium phosphate buffer, pH 7.2, or in this buffer containing 0.1 mM dithiothreitol (DTT) and 0.1 mM phenylmethylsulfonyl fluoride (PMSF).
CD spectroscopy. CD spectra were recorded on a J-600 (JASCO, Japan) apparatus. For calculation of the molar ellipticity per amino acid residue a value of the mean residue weight equal to 107.5 daltons was used. The secondary structure was calculated using methods from [9, 10].
RESULTS AND DISCUSSION
Expression and biological activity of gp9. Full-length gene 9 (see Fig. 1 for its nucleotide sequence) was cloned into the NcoI--BamHI sites of pET-23d(+) vector under the bacteriophage T7 promoter. As shown by SDS-PAGE analysis for E. coli BL21(DE3) proteins before and after inducing by IPTG, gp9 is expressed in large amounts (Fig. 2A, lanes 1 and 2a).
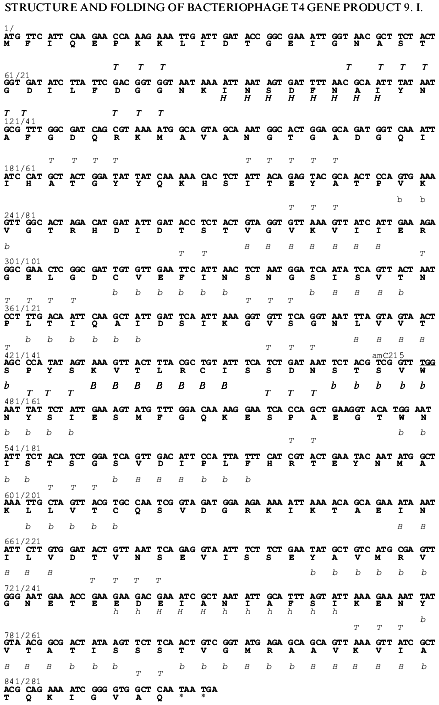
Fig. 1. Nucleotide and derived amino acid sequences of gp9 with the predicted secondary structure elements. The secondary structure elements: H, alpha-helix, high probability; h, alpha-helix, low probability; B, beta-structure, high probability; b, beta-structure, low probability.
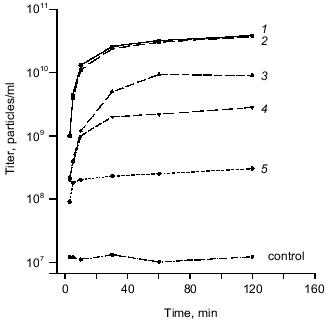
Biological activity of recombinant protein was studied in in vitro complementation assay using a cell lysate containing recombinant gp9. The data obtained from in vitro complementation assays are shown in Fig. 3 (curve 1). The titer of the phage increases by more than three orders of magnitude, what indicates correct folding and biological activity of the recombinant protein.Fig. 2. A, B) SDS-PAGE in 15% gel of the recombinant proteins after expression (a) and purification (b): 1) cell lysate before induction; 2) gp9; 3) NDelta20; 4) NDelta54; 5) NDelta167; 6) CDelta113. M, marker proteins.
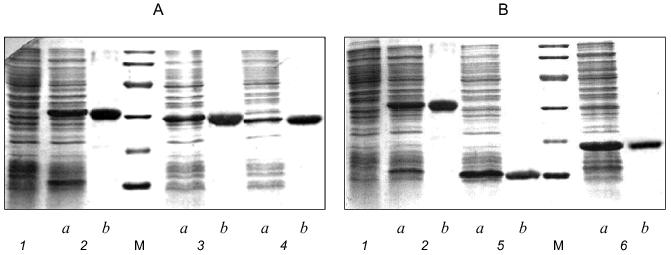
Purification and electrophoretic analysis of gp9. After sonication of the cells, the recombinant protein was precipitated by addition of saturated (NH4)2SO4 to the final concentration of 40-43%, and, after dissolving the precipitate in a small volume of 10 mM Tris-HCl buffer, pH 7.8, the protein was additionally purified by chromatography using hydroxyapatite. Electrophoretically pure fraction of gp9 was eluted in 10 mM sodium phosphate buffer, pH 7.8 (Fig. 2A, lane 2b).Fig. 3. Increase in a titer of the infective particles in in vitro complementation assay and effect of the deletion mutants of gp9 on the titer of infective particles: 1) gp9; 2) CDelta113; 3) NDelta20; 4) NDelta54; 5) NDelta167.
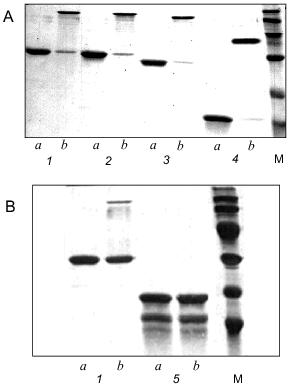
Electrophoretic analysis of the recombinant protein showed that the apparent mobility of a sample heated for 5 min at 95°C corresponded to the monomeric form of gp9 (Fig. 4A, lane 1a). However, for non-heated protein there is another band with apparent molecular weight of 94 kD (Fig. 4A, lane 1b). This result indicates that the native protein is a trimer resistant to SDS treatment at room temperature. It was believed in the past that gp9 existed in tetrameric form [11]. However, the trimeric nature of the gp9 confirmed by our studies corresponds to the trimeric nature of gp34, the proximal half of the LTF [12], and reflects the symmetry of interactions between these proteins.
Secondary structure predictions for gp9. Gene 9 was sequenced in our laboratory earlier [13]. The nucleotide and corresponding amino acid sequences of gp9 are shown in Fig. 1. The ALB program [14, 15] was used to calculate the probable secondary structure of gp9 based on its amino acid sequence. The predicted secondary structure elements are shown in Fig. 1. The relatively high content of beta-strands in central and C-terminal regions of the molecule as well as a large alpha-helical part in the N-terminus are notable.Fig. 4. A, B) SDS-PAGE of the purified recombinant proteins that were heated (a) and non-heated (b) before loading onto 15% gel: 1) gp9; 2) NDelta20; 3) NDelta54; 4) NDelta167; 5) CDelta113. M, marker proteins.
Secondary structure determination for gp9. The molar absorption coefficient for purified gp9 at 278.5 nm is 27,400 M-1·cm-1. CD spectra of recombinant gp9 are presented in Fig. 5. The CD spectrum in the far UV range (Fig. 5a) shows a single minimum at 216.3 nm ([theta]193.0 = 9900 deg·cm2/dmole), such CD spectra being characteristic of proteins with high beta-structure content and low alpha-helical content.
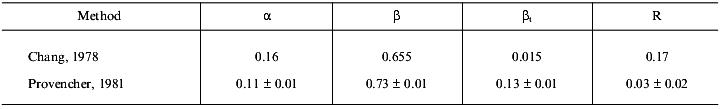
The CD spectrum of gp9 in the near UV range (Fig. 5b) shows a maximum at 242.8 nm ([theta]242.8 = 138 deg·cm2/dmole) and at 294.2 nm ([theta]294.2 = 29 deg·cm2/dmole) and several weak bands between 255 and 270 nm. These bands cannot be assigned with certainty. However, the weak bands at 255, 262, and 268 nm belong to side chains of phenylalanine of gp9 [16]. The near UV contribution to the protein CD spectra by aromatic side chain of tryptophan and tyrosine depends on the rigidity of the proteins, the interaction between the aromatic ring and the protein matrix, and the number of aromatic residues [16]. The CD intensity in this spectral range of disulfide of cysteine residues may be determined by the following three factors: the dihedral angle of the disulfide, the C--S--S bond angle, and the interaction with the protein matrix [16]. The positive molar ellipticity between 239 and 310 nm may originate from the disulfide(s). The independence of the CD spectra with regard to the presence of DTT in both the far and near UV range can indicate whether the disulfides are buried. The apparent maxima at 242.8 and 294.2 nm may originate from a cancellation of the positive CD signal of the disulfides and a negative CD signal from tyrosines and/or tryptophans at 284 nm. In either case, the intense CD spectrum in the near UV range is evidence for the existence of a rigid tertiary structure in the gp9 molecule. Results of calculations of secondary structure of gp9 by two different methods [9, 10] are shown in Table 1. The data suppose high content of beta-structure (65-73%) and low content of alpha-helices (11-16%) in the protein, this being in good agreement with the secondary structure predictions based on amino acid sequence [14, 15].Fig. 5. CD spectra of gp9 in the far (a) and near (b) UV ranges.
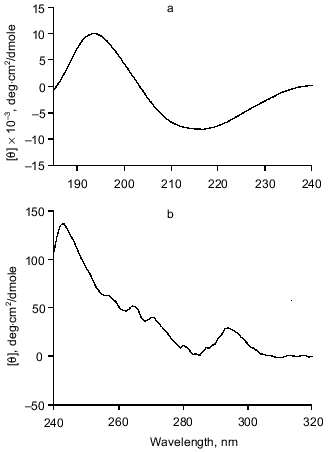
Table 1. Calculation of the secondary
structure of gp9
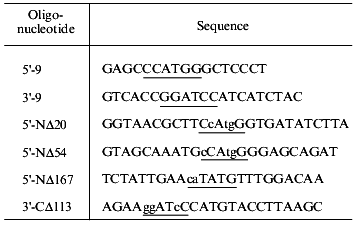
Production of the deletion mutants. Having the predicted secondary structure data, we constructed a set of vectors for expression of deletion mutants of gp9 to determine the role of the various parts of the molecule in its oligomerization. Fragments of gp9 of various lengths were produced via PCR using oligonucleotide primers with point mutations to generate convenient restriction sites (Table 2). Mutants with N-terminal truncations (NDelta20, NDelta54, and NDelta167) contain 268, 234, and 121 amino acid residues and have molecular weights of 29.5, 25.7, and 13.3 kD, respectively. The deletion mutant CDelta113 (molecular weight 19.1 kD) lacks 113 C-terminal amino acid residues. According to electrophoretic mobilities of the expressed proteins, their molecular weights are in good agreement with the calculated values (Figs. 2A and 2B, lanes 3a, 4a, 5a, 6a).
Table 2. Sequences of oligonucleotide
primers used for amplification of gene 9 and its fragments
The deletion fragments of gp9 were purified to apparent homogeneity as described for the full-length protein. The proteins were eluted from HAP using 10 mM sodium phosphate buffer, pH 7.8. The electrophoresis pictures of the purified proteins are shown in Figs. 2A and 2B (lanes 3b, 4b, 5b, 6b). The oligomeric organization of the proteins was determined by their resistance to SDS treatment as described for native gp9. Electrophoresis data are shown in Figs. 4A and 4B. As described for the full-length protein, the presence of additional intense bands was noted for non-heated samples of the protein mutants lacking consecutive N-terminal fragments corresponding to the trimeric forms (Fig. 4A, lanes 2b, 3b, 4b). However, there was no significant difference in electrophoresis pictures for the heated and non-heated samples of the CDelta113 mutant (Fig. 4B, lanes 5a and 5b), that is, the protein does not form trimers resistant to SDS treatment. It should be noted that mutant CDelta113 is not stable in solution, this being confirmed by the appearance of the bands corresponding to fragments with lower molecular weights (Fig. 4B, lanes 5a and 5b). These results indicate the trimeric nature of gp9 and its mutants with the N-terminal deletions and that the C-terminal part of the molecule is important for the trimerization of gp9.
We found that the deletion mutants of gp9 do not increase the titer of infective particles in in vitro complementation assay with 9--particles. However, these mutants had a dramatic effect on the subsequent complementation of the 9--particles by full-length gp9 molecules. In these experiments the 9--particles were incubated for 30 min at 30°C with cell lysates containing the deletion mutants, then the lysate containing the full-length protein was added. These data are shown in Fig. 3 (curves 2, 3, 4, 5). All mutants lacking the N-terminal fragments (NDelta20, NDelta54, and NDelta167) significantly decreased the titers of the infective particles (Fig. 3, curves 3, 4, 5). The mutant lacking the C-terminal fragment (CDelta113) did not influence the complementation of 9--particles by the full-length protein (Fig. 3, curve 2). Based on these results we conclude that the deletion mutants integrate into the baseplate or bind the LTF only in the presence of intact C-terminal part of the molecule.
We developed a system for expression and purification of biologically active recombinant gp9 and its deletion mutants. The native protein is a trimer resistant to SDS treatment. CD spectroscopy data and the secondary structure predictions indicate that gp9 is a beta-structural proteins. The C-terminal part of the molecule is important for assembly of a biologically active trimer.
We recently obtained crystals of recombinant gp9 [17], and now we are finishing the determination of its spatial structure using protein crystallographic techniques.
This study was supported by grants from the Russian Foundation for Basic Research (Nos. 97-04-49708 and 99-04-48430), “Universities of Russia” program (No. 2608), and Howard Hughes Medical Institute (No. 75195-520803).
REFERENCES
1.Coombs, D. H., and Arisaka, F. (1994) in
Molecular Biology of Bacteriophage T4 (Karam, J. D., ed.)
American Society for Microbiology, Washington, D.C., pp. 259-281.
2.Goldberg, E., Grinius, L., and Leteller, L. (1994)
in Molecular Biology of Bacteriophage T4 (Karam, J. D., ed.)
American Society for Microbiology, Washington, D.C., pp. 347-356.
3.Crowther, R. A. (1980) J. Mol. Biol.,
137, 159-174.
4.Studier, F. W., Rosenberg, A. N., Dunn, A. H., and
Dubendorff, J. W. (1990) Meth. Enzymol., 185, 60-89.
5.Laemmli, U. K. (1970) Nature, 227,
680-685.
6.Edgar, R. S., and Wood, W. B. (1966) Proc. Natl.
Acad. Sci. USA, 55, 498-505.
7.Venyaminov, S. Y., and Gogia, Z. V. (1982) Eur.
J. Biochem., 126, 299-309.
8.Jaenicke, L. (1974) Anal. Biochem.,
61, 623-627.
9.Chang, C. T., Wu, C.-S. C., and Yang, J. T. (1978)
Anal. Biochem., 91, 13-31.
10.Provencher, S. W., and Glockner, J. J. (1981)
Biochemistry, 20, 33-37.
11.Kikuchi, Y., and King, J. (1975) J. Mol.
Biol., 99, 645-672.
12.Cerritelli, M. E., Simon, M. N., Vaca, M.,
Conway, J. F., and Steven, A. C. (1994) Proc. XIII Int. Congr. on
Electron Microscopy, Paris.
13.Prilipov, A. G., Selivanov, N. A., Efimov, V. P.,
Marusich, E. I., and Mesyanzhinov, V. V. (1989) Nucleic Acids
Res., 17, 3303.
14.Finkelstein, A. V. (1975) Dokl. Akad. Nauk
SSSR, 223, 744-747.
15.Ptitsyn, O. B., and Finkelstein, A. V. (1983)
Biopolymers, 22, 15-25.
16.Strickland, E. H. (1974) CRC Crit. Rev.
Biochem., 2, 113-175.
17.Strelkov, S. V., Zurabishvili, T. G., Nepluev, I.
V., Efimov, V. P., Isupov, M. N., Harutunyan, A. G., and Mesyanzhinov,
V. V. (1993) J. Mol. Biol., 234, 493-495.