REVIEW: Troponin: Structure, Properties, and Mechanism of Functioning
V. L. Filatov1*, A. G. Katrukha2, T. V. Bulargina1, and N. B. Gusev3
1Department of Bioorganic Chemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-2788; E-mail: filatov@soil.msu.ru2Moscow Scientific-Research Institute of Medical Ecology, Simferopolskii Bulvar 8, Moscow, 113149 Russia; fax: (095) 939-2788; E-mail: katrukha@soil.msu.ru
3Department of Biochemistry, School of Biology, Lomonosov Moscow State University, Moscow, 119899 Russia; fax: (095) 939-3955; E-mail: gusev@gusev.bio.msu.su
* To whom correspondence should be addressed.
Received March 22, 1999; Revision received April 28, 1999
This review discusses the structure and properties of the isolated components of troponin, their interaction, and the mechanisms of regulation of contractile activity of skeletal and cardiac muscle. Data on the structure of troponin C in crystals and in solution are presented. The Ca2+-induced conformational changes of troponin C structure are described. The structure of troponin I is analyzed and its interaction with other components of actin filaments is discussed. Data on phosphorylation of troponin I by various protein kinases are presented. The role of troponin I phosphorylation in the regulation of contractile activity of the heart is analyzed. The structural properties of troponin T and its interaction with other components of thin filaments are described. Data on the phosphorylation of troponin T are presented and the effect of troponin T phosphorylation on contractile activity of different muscles is discussed. Modern models of the functioning of troponin are presented and analyzed.
KEY WORDS: troponin, tropomyosin, actin, regulation of muscle contraction
In the 1960s it was hypothesized that the contraction of striated muscle is regulated by a special protein complex located on actin filaments; this complex was called native tropomyosin [1]. It has been shown that native tropomyosin consists of two parts, namely, tropomyosin, already described by Bailey in 1946 [2], and troponin. The first fundamental investigations of troponin were performed in the laboratories of S. Ebashi [1], J. Gergely [3], and S. V. Perry [4]. Troponin consists of three components, each of which performs specific functions. Troponin C binds Ca2+, troponin I inhibits the ATPase activity of actomyosin, and troponin T provides for the binding of troponin to tropomyosin.
The recent decades have been marked by the development of site-directed mutagenesis and sophisticated physical methods. These approaches have provided new data on the structure of troponin and suggested new ideas on the functioning of troponin in muscles. In addition to fundamental interest, investigation of troponin is also important from the practical point of view. For example, it is important to develop new drugs increasing the affinity of troponin C to Ca2+, and in this way enhancing the contractile activity of the myocardium [5]. The effects of various hormones on troponin phosphorylation and the effects of this process on the regulation of contractile activity of striated and cardiac muscle is under detailed investigation [6, 7]. Also, components of troponin are widely used as biochemical markers of various heart injuries (see for example [8-10]).
The main goal of this review is to summarize recent experimental data on the structure, properties, and mechanism of functioning of the troponin complex. These data are interesting not only from the theoretical point of view; they can also be used by investigators studying troponin for practical purposes. At the beginning we will describe some properties of the isolated troponin components, then we will analyze their interaction and the mechanism of their coordinate functioning.
I. TROPONIN C
General Data on the Structure and Parameters of Ca2+ Binding
Troponin C is the Ca2+-binding component of troponin. The primary structures of skeletal and heart troponin from various species of mammals, birds, and some invertebrates have been described in the literature [11]. There are no less than two genes for troponin C in tissues if higher animals. One gene codes the isoforms characteristic for fast skeletal muscle fibers, whereas the second gene codes the isoform characteristic for slow skeletal fibers and heart [11, 12]. All isoforms of troponin C have low isoelectric points, and they are highly homologous. Troponin C contains four motifs having helix--loop--helix structures. This conservative feature was first found in the three-dimensional structure of parvalbumins (Ca2+-binding proteins from fish muscle) and was called the EF-hand [11]. The family of EF-hand proteins is now very large and contains various intracellular proteins having high affinity for Ca2+; they function either as Ca2+-buffers (calbindin, parvalbumins) or as Ca2+-dependent triggers (calmodulin, troponin C, myosin light chains, etc.) [13].
The typical EF-hand consists of a 12-membered loop which is flanked on both sides by alpha-helices containing 12-14 amino acid residues. Six residues located in positions 1, 3, 5, 7, 9, and 12 of the 12-membered loop are directly involved in Ca2+ binding. The cation is bound to oxygen atoms belonging to the carboxyl or hydroxyl groups of amino acid residues, to the oxygen of the carbonyl group of the peptide bond, or to the oxygen of a water molecule fixed inside the loop. The conservative Glu residue located in the twelfth position of the loop donates two oxygen atoms of the carboxyl group for the binding of Ca2+. Seven oxygen atoms are located in the vertexes of a pentagonal bipyramid, and the polypeptide chain winds around the cation [13, 14]. To some extent the primary structure of the loop determines the parameters of Ca2+ binding. Therefore the primary structure of the Ca2+-binding sites is highly conservative [11]. However, the specificity and affinity is determined not only by the primary structure of the loop itself but is also dependent on the primary structure of the helices flanking the Ca2+-binding loop [15], the helices located in the vicinity of the Ca2+-binding loop [16], as well as the interaction between neighboring loops [17].
Three-Dimensional Structure of Troponin C
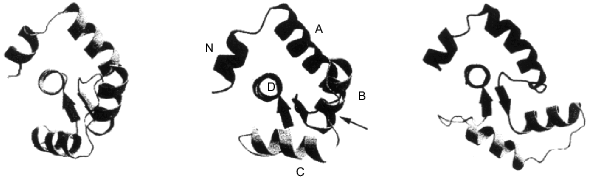
The first data on the three-dimensional structure of troponin C appeared in 1985, and a detailed structure with 2 Å resolution was published in 1988 [18, 19]. So far, all attempts to crystallize troponin C in the absence of Ca2+ have been unsuccessful. However, it is possible to obtain crystals of troponin C with the two C-terminal [18, 19] or all four (two N-terminal and two C-terminal) Ca2+-binding sites [20] saturated with Ca2+. The X-ray crystallographic data indicate that troponin C has a dumbbell-like form. Two globular domains each contain two Ca2+-binding sites. A handle formed by a long central alpha-helix connect these two globular domains (Fig. 1). Thus, according to the X-ray data troponin C has a rather symmetric, elongated form. Recently published high resolution NMR data [21] principally agree with the results of X-ray crystallography. However, according to the X-ray crystallography, the central **alpha-helix is extended and rigid, whereas the NMR data indicate that the central helix is partially melted and the hinge region formed in the middle of the central helix provides for high relative mobility between the two globular domains (Fig. 2). The NMR data agree with earlier published results on troponin C structure obtained by low angle X-ray scattering [22].
Fig. 1. Crystal structure of rabbit skeletal troponin C completely saturated with Ca2+ [20] (PDB code 1TN4, 2TN4). The figure was produced by the RasMol program [156].

Numerous experiments performed by various methods also indicate that the two globular domains can approach each other [23]. Thus, depending on conditions, isolated troponin C will assume either a symmetrical extended dumbbell-like form or a rather compact form with its globular domains closely approaching each other. The troponin C molecule consists of three main components, namely, the two globular domains and the central alpha-helix linking these domains. Let us analyze each of these elements separately.Fig. 2. Structure of completely Ca2+-saturated troponin C in solution obtained by high resolution NMR [21] (PDB code 1TNW). The two globular domains of troponin C can move with respect to each other due to partial melting of the central alpha-helix. The figure was drawn by the RasMol program [156].

N-Terminal globular domain of troponin C. The N-terminal domain contains two Ca2+-binding sites. The affinity of these sites (Ka ~ 106 M-1) is lower than affinity of the Ca2+-binding sites located in the C-terminal domain of troponin C [24], although the N-terminal sites possess higher specificity. These sites practically do no bind Mg2+, and therefore they are designated as Ca2+-specific low affinity Ca2+-binding sites. It is supposed that the low affinity of these sites is due to the fact that these sites contain in the third position of the Ca2+-binding loop Gly or Ser. These residues are less effective in coordinating Ca2+ than the Asp located in the homologous position of the third and the fourth Ca2+-binding site. The presence of Asp in position 3 of the third and the fourth Ca2+-binding sites also provides for binding of Mg2+ in the C-terminal domain of troponin C [20]. The primary structure of the N-terminal domain of cardiac troponin C is different from that of the skeletal muscle troponin C [11]. Actually, the second Ca2+-binding site of cardiac troponin C retains the specificity and affinity characteristic of the second Ca2+-binding site of skeletal troponin C [25], whereas the first cation-binding site of cardiac troponin C contains an insertion and nonconservative replacements and is therefore unable to bind bivalent cations [11].
The N-terminal domain of troponin C contains five alpha-helices. In addition to helices A and B flanking the first Ca2+-binding site and helices C and D flanking the second Ca2+-binding site, the N-terminal domain of troponin C contains an additional alpha-helix located at the very N-terminus of the protein. This so-called N-helix plays an important role in the proper alignment of the whole N-terminal domain of troponin C. Removal of the N-helix leads to decrease in the affinity of the cation-binding sites and hampering of the regulatory properties of troponin C [26]. Of a special importance is Arg-11, forming salt bridges with Glu-16 (helix A) and Glu-76 (helix D). The point mutation R11A leads to destabilization of the central helix and decreased ability of troponin C to regulate the interaction of myosin with actin [27].
The sequence of processes occurring in the N-terminal domain during saturation by Ca2+ has been followed using site directed mutagenesis. In skeletal troponin C the first Ca2+-binding site has lower affinity than the second site [17, 28, 29]. This is due to the presence of three consecutive Gly residues in the structure of the first Ca2+-binding sites. These Gly residues provide extreme flexibility, and therefore the first Ca2+-binding loop cannot bind Ca2+ tightly.
C-Terminal globular domain of troponin C. The C-terminal domain contains two sites which are able to bind both Mg2+ (Ka ~ 2·103 M-1) and Ca2+ (Ka ~ 2·107 M-1) [24]. These sites are designated as high-affinity Ca2+,Mg2+-sites. During relaxation at low concentration of Ca2+ in myoplasm the C-terminal sites of troponin C are saturated with Mg2+. The rate constant of dissociation of Mg2+ from these sites is rather low (koff = 8 sec-1); therefore, during initiation of contraction and increase in Ca2+ concentration in myoplasm, magnesium ion is only very slow replaced by calcium ion. It is concluded that the C-terminal sites of troponin C cannot directly participate in the regulation of muscle contraction. These sites seem to play important structural role providing for fixation of troponin C to other components of the thin filament [30, 31]. The experimental data confirm this conclusion. The point mutations with replacement of the first residue of the third and/or the fourth Ca2+-binding loop (Asp) by Ala [30, 31] result in complete blocking of Ca2+ binding in the third and/or the fourth Ca2+-binding site(s). Nevertheless, these mutated proteins were incorporated into actin filaments which were previously depleted of exogenous troponin C. These mutants effectively regulated the tension of skinned muscle fibers [31]. These mutants do not differ from the wild type troponin C in their ability to regulate the interaction of myosin with actin but interact with the proteins of thin filaments with lower affinity [31]. The special role of the third cation-binding site of troponin C is conferring of the structural stability of troponin C inside the thin filament [31].
Binding of Ca2+ in the N-terminal domain is accompanied by reorientation of already formed elements of the secondary structure [23]. In contrast, binding of Ca2+ in the C-terminal domain induces formation of additional alpha-helices and formation of a short beta-sheet between the third and the fourth cation-binding sites [32]. During relaxation the C-terminal sites of troponin C are saturated with Mg2+, which also affects the structure of the C-terminal domain of troponin C. Now there is no doubt that the cation-binding sites of the C-terminal domain play an important role in the maintenance of the whole structure of troponin C and in its incorporation into thin filaments.
The central alpha-helix of troponin C. The globular domains of troponin C are connected by a long alpha-helix (Fig. 1). It was hypothesized that this helix plays an important role in transduction of conformational signals between the two Ca2+-binding globular domains of troponin C [33]. A set of troponin C mutants with deletions, insertions, and point mutations in the central helix was created for testing this hypothesis. Removal of three, four, or seven amino acid residues in the central helix has no significant effect on the properties of troponin C [34]. These mutations are not accompanied by significant rotation of the globular domains with respect to each other. Removal of two residues of the central helix results in rotation of globular domains by 180° with respect to each other. This reorientation slightly changed the ability of troponin C to regulate the interaction of myosin with actin [35]. Recently, mutants with deletion of 11 and even 12 residues of the central helix of troponin C were described in the literature [34]. These dramatic changes in the length of the central helix have no effect on the parameters of Ca2+ binding. However, these mutants were ineffective in regulation of actomyosin ATPase. These mutants tend to dimerization, and in the dimeric state they were able to regulate the interaction of myosin and actin.
Mutants of troponin C with lengthened central helix are also described in the literature. Elongation of the central helix was achieved by insertion of additional turns of the alpha-helix, insertion of nine Pro residues or by insertion of nine residues tending to form coil [36]. Incorporation of nine Pro residues or coil-forming residues results in markedly decreased ability of troponin C to regulate the actomyosin ATPase activity.
Thus, we conclude that the central helix forms a semi-flexible hinge connecting the two globular domains of troponin C. In addition, the central helix seems to participate in the conduction of signals between the globular domains of the protein [30].
Ca2+-Induced Conformational Changes of Troponin C
Initiation of muscle contraction begins with the saturation of the regulatory sites of troponin C by Ca2+. Therefore, to understand the processes underlying the regulation of muscle contraction, it is important to follow the Ca2+-induced conformational changes of troponin C. X-Ray crystallography has provided data on the structure of troponin C with the N-terminal sites free of Ca2+ and the C-terminal sites saturated with Ca2+ [37]. In this case the structure of the N-terminal domain is significantly different from that of the C-terminal domain. The main difference is that in the C-terminal domain (saturated with Ca2+) helices F, G, and H flanking the Ca2+-binding loops are oriented practically perpendicular to the central helix, whereas in the N-terminal domain (free of Ca2+) helices B and C are nearly parallel to the central helix. Herzberg, Moult, and James [38] hypothesized that after saturation with Ca2+ the helices of the N-terminal domain would adopt the same conformation as the helices of the C-terminal domain. Thus, it was postulated that binding of Ca2+ by the regulatory centers would lead to the movement of helices B and C with respect to the central helix and their orientation nearly perpendicular to it. Reorientation of helices would expose some of the hydrophobic residues located in the region of the contact of the central helix and helices B and C. Exposure of these residues might play a significant role in the formation of tight contacts between troponin I and troponin C.
Each point of this hypothesis was checked by point mutation of certain residues in troponin C. The interaction of helices C and D was enhanced by introduction of oppositely charged residues and formation of electrostatic contacts [39]. The movement of helices B and C against the central helix of troponin C was restricted by incorporation of a disulfide bond cross-linking the linker between helices B and C to the central helix (helix D) of troponin C [40]. Any way of immobilization of helices B and C against the central helix results in a significant decrease in the affinity to Ca2+ and nearly complete loss of ability to regulate actomyosin ATPase or tension of skinned fibers. As already mentioned, the movement of helices B and C against the central helix is accompanied by exposure of hydrophobic residues, which is an energetically unfavorable event. Mutations V45T, M46Q, M48A, L49T, and M82Q decrease the hydrophobic contacts between helices B and D. These mutations facilitate movement of helix B with respect to helix D and are accompanied by increased affinity of the regulatory sites of troponin C to Ca2+ [41]. All these results are in complete agreement with the hypothesis of Herzberg, Moult and James [38].
The hypothesis was conclusively confirmed by X-ray crystallography [20] and NMR spectroscopy [21] of troponin C completely saturated with Ca2+. Indeed, in this case the orientations of the helices in the N- and C-terminal domains of troponin C were practically identical. In the absence of Ca2+, the Ca2+-binding sites are in the so-called closed configuration. In this state hydrophobic residues of the helices flanking the Ca2+-binding sites form contacts with each other and are shielded from the solvent. In this state the helices flanking the Ca2+-binding sites interact with the central helix. After saturation with Ca2+, the helices belonging to the Ca2+-binding sites move farther apart from the central helix, hydrophobic residues become exposed to the solvent, and the Ca2+-binding sites change to their so-called opened conformation.
All the events described so far are characteristic for skeletal troponin C, all centers of which are able to bind Ca2+. As already mentioned, cardiac troponin C, due to nonconservative replacements in the first site, has lost its ability to bind Ca2+ in this center [11]. Nevertheless, cardiac troponin C regulates the interaction of myosin with actin. This means that the second site plays a major role in the regulation of cardiac contraction. Indeed, mutation of Glu-65, thus switching off of the second cation-binding site completely, abolishes the regulatory properties of cardiac troponin C [42]. This means that the second site of cardiac troponin C plays an especially important role in the regulation of contraction.
Simple switching off of the first site (mutations D27A and D29A) of skeletal troponin C is not enough to make the properties of skeletal troponin C similar to those of cardiac troponin C [43]. Only replacement of the first 41 residues of skeletal troponin C by the corresponding residues of cardiac troponin C makes the properties of this chimerical protein similar to those of cardiac troponin C [43].
Recently published results directly show that the conformational changes of the N-terminal domain of cardiac troponin C are different from the corresponding changes in the structure of skeletal troponin C. In both cases the second cation-binding site has higher affinity and is saturated with Ca2+ before the first Ca2+-binding site [17, 28, 44]. Only after saturation of the second site does Ca2+ bind to the first site. During this process the side chain of the residue located in the twelfth position of the first Ca2+-binding site (Glu-41 in skeletal troponin C) is reoriented. Only after this reorientation all helices start to move, and the N-terminal domain is switched from the closed to the open conformation. Cardiac troponin C (or skeletal troponin C with mutation E41A) is unable to bind Ca2+ in the first loop. Therefore, after binding of Ca2+ in the second loop, there is no change in the orientation of the twelfth ligand of the first Ca2+-binding loop, and the N-terminal domain remains in the so-called closed conformation [28, 45, 46]. As shown in Fig. 3, saturation of the N-terminal domain of skeletal troponin C is accompanied by significant movement of helices B and C with respect to helices N, A, and D (the left and right schemes in Fig. 3). At the same time, Ca2+ binding by the N-terminal domain of cardiac troponin C does lead to significant changes in the structure (left and central schemes in Fig. 3). Ca2+ binding leads to exposure of hydrophobic residues of the N-terminal domain of skeletal troponin C [44] and does not affect accessibility of homologous hydrophobic residues of cardiac troponin C [46]. Thus, with the isolated proteins, Ca2+ evokes completely different changes in the structure of cardiac and skeletal troponin C.
Despite these differences, both isoforms of troponin C regulate muscle contractile activity. There are several explanations for this discrepancy. It is known that cardiac troponin C and troponin I interact with each other more weakly than the corresponding components of skeletal muscle [47]. This is possible if the sites of interaction are different for troponin components of the heart and skeletal muscle. Another explanation is based on the assumption that cardiac troponin I evokes structural changes of cardiac troponin C. These structural changes might facilitate reorientation of the helices in the N-terminal domain of troponin C such that Ca2+ binding would lead to the opening of the N-terminal domain [45]. Some indirect support for this suggestion is the fact that the interaction with troponin I changes the environment of Met-81 of cardiac troponin C [48].Fig. 3. Structure of the N-terminal domain of troponin C in different states [45] (PDB code 2CTN). Left, the N-terminal domain of skeletal troponin C free of Ca2+. In the center, the N-terminal domain of cardiac troponin C with the second site saturated with Ca2+. Right, the N-terminal domain of skeletal troponin C with the first and the second sites saturated with Ca2+. The helices are marked by capital letters. The arrow marks the position of Glu-40 of cardiac troponin C.
Summing up, we conclude that Ca2+ binding is accompanied by large conformational changes in the structure of troponin C. These changes can be transferred to the other components of troponin complex.
Interaction of Troponin C with Other Components of the Troponin Complex
Troponin C interacts with troponin I, and the strength of this interaction is sharply increased in the presence of Ca2+. So far there are no X-ray crystallographic data on the structure of the binary complex of troponin C and troponin I. Data from low-angle X-ray diffraction indicate that troponin I winds around the dumbbell-like molecule of troponin C [49]. Recently published X-ray crystallographic data obtained on the complex of troponin C with a short peptide of troponin I (residues 1-47) show that troponin C bent in the middle of its central alpha-helix interacts via its C-terminal domain with this peptide of troponin I [50]. It is supposed that the central and the C-terminal parts of troponin I may interact with the N-terminal globular domain of troponin C, which is bent in the middle of its central alpha-helix [50]. It is still unknown how troponin components interact inside the whole troponin complex, but both models published so far indicate that both proteins interact through multiple contacts covering a large part of their surfaces. The polypeptide chains of troponin C and troponin I are oriented antiparallel to each other [51, 52]. The central helix and both globular domains of troponin C form contacts with different sites of troponin I [53, 54]. This interaction is very dynamic, and the strength of interaction and formation and breaking of certain contacts depend on the presence of Ca2+ [52].
The interaction of troponin C with troponin T has been investigated in less detail. Nevertheless, it is known that under certain conditions these proteins can interact with each other [55, 56]. The N-terminal domain of troponin C seems to play the major role in the interaction with troponin T. Recently published data indicate that conservative acidic residues of troponin C (Glu-53, Glu-54 and Glu-85, Glu-86) are especially important for the interaction with troponin T [56a]. This interaction is enhanced in the presence of Ca2+ and may play a role in the regulation of muscle contraction [56].
II. TROPONIN I
Structure and Properties of Troponin I
Troponin I is responsible for inhibition of the actomyosin ATPase [3]. Three isoforms of troponin I have been described for striated muscle. Two isoforms are characteristic for skeletal fibers (for fast and slow skeletal fibers) and one isoform for cardiac muscle [57, 58]. Troponin I consists of 181-211 amino acid residues, and the cardiac isoform is larger due to the presence of an additional approximately 30-membered N-terminal peptide [58]. Troponin I is a polar protein with an excess of positively charged residues; therefore, its calculated pI is approximately 9.9.
The troponin I isoforms are coded by three different genes. It is known that the gene of human cardiac troponin I is located on the 19th chromosome [59, 60] and consists of eight exons. Expression of troponin I genes is dependent on the stage of ontogenesis [61]. Both the cardiac and the slow skeletal isoforms of troponin I are expressed in the heart of the human fetus. After birth the expression of the slow skeletal isoform is blocked, whereas the expression of the cardiac isoform is enhanced. The final result of this switching is exclusive expression of the cardiac isoform of troponin I by the ninth month of life [62]. Since the level of mRNA corresponds to the level of protein synthesized, the regulation of isoform expression occurs on the level of translation.
Interaction of Troponin I with Other Components of the Thin Filament
Inhibitory sites of troponin I and the interaction of troponin I with actin and tropomyosin. As already mentioned, the main function of troponin I is the inhibition of actomyosin ATPase activity [3]. This inhibition is enhanced in the presence of tropomyosin and is completely abolished in the presence of fully Ca2+-saturated troponin C [4, 58]. To understand the mechanism of functioning of troponin I, it is important to analyze its interaction with actin, tropomyosin, and troponin C.
Different fragments of troponin I have been produced in order to map the sites of interaction of troponin I with other components of the thin filament. Among fragments of skeletal troponin I, a short peptide consisting of residues 96-116 retains the ability to inhibit actomyosin ATPase activity [63]. This peptide inhibits the contraction of skinned cardiac and skeletal fibers depleted of endogenous troponin I [58, 64]. In addition, this peptide retains other important properties of intact troponin I, and it interacts with actin and troponin C [63]. Later, a second inhibitory peptide was found in the structure of troponin I. This inhibitory region consists of residues 128-148 [52]. The NMR data indicate that the first inhibitory peptide (residues 96-116) interacts with residues 1-7 and 19-44 of actin [65], i.e., it recognizes residues involved in the interaction of actin with myosin. The location of the second inhibitory peptide on the surface of actin is still unknown (Figs. 4 and 5). The primary structure of the two inhibitory peptides (residues 101-114 and 121-132) reveals some homology [66]. The role of various residues in the structure of the first (main) inhibitory peptide has been analyzed in detail. Residues 104-115 contain the minimal sequence retaining inhibitory activity, and residues 105 and 114 play crucial roles [67]. The inhibitory peptides of cardiac and skeletal troponin I have rather conservative primary structure, but, nevertheless, skeletal troponin I is more effective than its cardiac counterpart in inhibition of actomyosin ATPase activity and contraction of skinned fibers [68]. It is supposed that this difference is due to the single nonconservative replacement in the inhibitory peptide of cardiac troponin I. The inhibitory peptide of skeletal troponin I contains Pro in positions 109 and 110, whereas cardiac troponin I contains Pro and Thr in the corresponding positions. The inhibitory peptide of skeletal troponin I, 96NQKLFDLRGKFKRPPLRRVRM116, forms two amphiphilic alpha-helices rigidly fixed in the form of a hairpin by the dipeptide Pro-109--Pro-110. In the case of cardiac troponin I, the two alpha-helices of the inhibitory peptide have greater freedom and, therefore, less effectively inhibit the actomyosin ATPase activity. A synthetic inhibitory peptide of skeletal troponin I with the replacement P110G is also characterized by increased mobility of the two alpha-helices. This leads to its decreased inhibition of the actomyosin ATPase activity [67, 69].
Fig. 4. Scheme of the interaction of troponin I (TnI) with thin filament proteins. Troponin C (TnC) is shown as a dumbbell-like with marked globular N- and C-terminal domains. Actin is shown as an ellipse. The sites interacting with inhibitory peptides of troponin I are marked. Troponin T (TnT) is shown in the form of a comma. The borders of the sites of interaction are indicated by numbers.
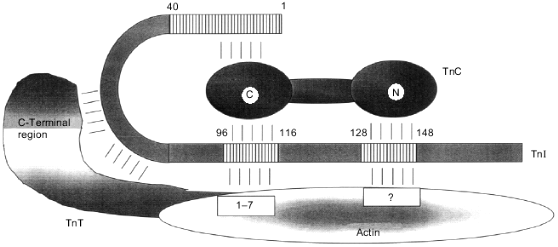
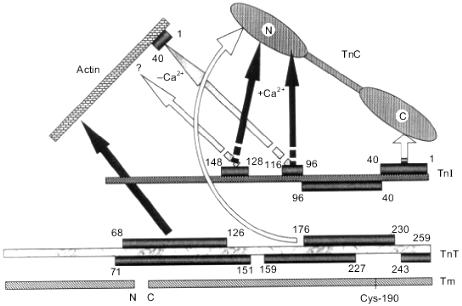
Summing up, we conclude that troponin I contains at least two inhibitory sites. Both of these sites are basic. The first (main) inhibitory site (residues 96-116) has a hairpin form formed by two rigidly fixed with respect to each other alpha-helices.Fig. 5. Scheme of interactions of thin filament proteins. Each protein is shown as a segment. Residue numbers shown in rectangles which face each other or are connected by arrows indicate the sites of interaction between partners. The positions of the N- and C-termini of the proteins are indicated. Arrows labeled +Ca2+ and -Ca2+ indicate the interactions which are enhanced in the presence or in the absence of Ca2+, respectively. Tm, tropomyosin; TnT, troponin T; TnI, troponin I; TnC, troponin C.
The inhibitory activity of troponin I is significantly increased in the presence of tropomyosin [4, 58]. This fact suggests a direct interaction of troponin I and tropomyosin. This suggestion was proved experimentally [70, 71], although the sites of troponin I--tropomyosin interaction are still not exactly mapped.
Interaction of troponin I with troponin C. As already mentioned, the inhibitory effect of troponin I can be reversed by troponin C. This is possible because these two proteins tightly interact with each other. In the absence of Ca2+ the inhibitory sites of troponin I interact with actin, whereas in the presence of Ca2+ these sites interact with troponin C.
The orientations of the polypeptide chains of troponin I and troponin C are antiparallel [48, 72]. With this orientation the first (main) inhibitory peptide (residues 96-116) of troponin I may interact both with the N- and C-terminal domains of troponin C [53, 54, 74] or may be located close to the central alpha-helix and the C-terminal domain of troponin C [74, 75]. Replacement of Pro-110 with Thr or Gly in the inhibitory peptide of troponin I leads to a decrease in its inhibitory action [67, 69]. This mutation also affects the interaction of the inhibitory peptide of troponin I and troponin C. The intact inhibitory peptide increases the affinity of the Ca2+-binding sites of troponin C to Ca2+, whereas the mutant peptide with replacement P110G does not affect the Ca2+-binding properties of troponin C [76]. Thus, depending on conditions, the same inhibitory peptide of troponin I may interact with actin (and inhibit ATPase activity of actomyosin) as well as with troponin C (and this will challenge the inhibitory action of troponin I).
The contact sites of troponin I and troponin C have been described in detail (Fig. 5). Without going into the details, we note that the N-terminal part of troponin I (residues 1-40) interacts with the C-terminal globular domain of troponin C [52, 66]. The first (main) inhibitory peptide of troponin I (residues 96-116) can interact both with the central helix and the two globular domains of troponin C [53, 54, 73-75]. The C-terminal part of troponin I (residues 116-148) interacts with the N-terminal domain of troponin C [66, 77]. (Very recently published results indicate that residues 138-148 do not appear to directly interact with troponin C, therefore the C-terminal part of troponin I interacting with troponin C should be restricted to residues 116-137 [77a]). In the absence of Ca2+, troponin C weakly interacts with troponin I, which binds to actin through its inhibitory and actin-binding sites (residues 96-116 and probably 140-148) and inhibits actomyosin ATPase activity. Binding of Ca2+ to the regulatory sites of troponin C enhances the interaction of troponin C with the region of troponin I including residues 116-131 and of dissociation of the main inhibitory site of troponin I (residues 96-116) from actin. This results in disinhibition of the actomyosin ATPase activity.
The model described explains the interaction of troponin I and troponin C inside the skeletal troponin complex. As already mentioned, the primary structure of cardiac troponin I and troponin C differ from those of the skeletal muscle counterparts. Therefore, at present it is difficult to exclude differences in the interaction of the C-terminal fragment of troponin I and troponin C in cardiac muscle [78].
All the models described are hypothetical and are based on experiments performed on deletion mutants and proteolytic fragments of the components of troponin. So far, no X-ray crystallographic data of the whole troponin complex or of binary complexes of troponin I and troponin C are available. Low-angle X-ray diffraction data indicate that the binary troponin I--troponin C complex has an elongated form and that troponin I winds around the extended troponin C molecule. The centers of mass of both proteins coincide, and both proteins are located symmetrically with respect to each other [49, 79]. Taking into account the antiparallel orientation of the polypeptide chains of troponin C and troponin I, this model does not contradict any earlier described results. However, recently published results [50, 66, 80] favor a more compact alignment of the troponin components.
Interaction of troponin I with troponin T. The tight interaction of troponin I and troponin T has been demonstrated bygel-filtration, circular dichroism, and chemical cross-linking. The association constant for troponin I and troponin T was estimated to be close to 8.5·106 M-1 [81]. The strength of the interaction depends on the reduction of Cys-48 and Cys-64 of troponin I [82]. Deletion of the first 57 residues of troponin I does not affect its interaction with troponin T [83]. Troponin T affects the accessibility of Lys-70 of troponin I to chemical modification [84]. Taking into account all these experimental facts, we conclude that the N-terminal part of troponin I (residues 40-96) participates in the interaction with troponin T in the binary complex. Chemical cross-linking of the whole troponin complex also indicates that troponin I and troponin T are in close vicinity [85, 86]. The interaction of troponin I and troponin T inside the whole troponin complex depends on Ca2+. It is supposed that a coiled coil formed by alpha-helix containing residues 53-106 of troponin I and alpha-helix containing residues 205-255 of troponin T provides for the troponin I--troponin T interaction [51, 87]. Thus, the site of troponin T binding seems to be located between residues 40-96 of troponin I, i.e., it is squeezed between residues 1-40 and 96-148 of troponin I involved in the interaction with troponin C [51]. This location of the binding sites provides for the transfer of conformational signal induced by binding of Ca2+ to troponin C through troponin I to troponin T [51] and tropomyosin.
Phosphorylation of Troponin I
Protein--protein interactions inside the troponin complex are controlled not only by Ca2+, but can be modulated by troponin I phosphorylation as well. Phosphorylation is especially characteristic for cardiac troponin I.
Under in vitro conditions troponin I can be phosphorylated by three protein kinases, i.e., Ca2+-phospholipid-dependent protein kinase (protein kinase C, PKC), cGMP-dependent protein kinase (protein kinase G, PKG), and cAMP-dependent protein kinase (protein kinase A, PKA).
Phosphorylation by Ca2+-phospholipid-dependent protein kinase (protein kinase C, PKC). Unfractionated protein kinase C preparations phosphorylate Ser-144 and also 43, 45, 23, and 24 of isolated cardiac troponin I and of troponin I inside the whole troponin complex [88]. More detailed investigations [89, 90] revealed that different isoforms of PKC phosphorylate different residues in troponin I. The delta-isoform of PKC predominantly phosphorylates Ser-23 and 24 (as well as Ser-43 and -45). The alpha- and epsilon-isoforms of protein kinase C predominantly phosphorylate Ser-43 and -45 of troponin I, whereas the zeta-isoform of PKC poorly phosphorylates troponin I. Addition of phorbol esters or stimulation of alpha1-adrenoreceptors (i.e., under conditions stimulating PKC) leads to phosphorylation of the same sites which are phosphorylated by PKC in vitro [91]. However, perfusion of guinea pig hearts with solutions containing agonists of alpha1-adrenoreceptors or phorbol esters was not accompanied by an increase in the level of troponin I phosphorylation [92]. The data of Noland et al. [88] indicate that the phosphorylation of troponin I by PKC results in a decrease in actomyosin ATPase activity without significant change in its dependence on Ca2+ concentration. Thus, if PKC phosphorylates troponin I in vivo, this will lead to a decrease in ATPase activity of myofibrils and probably to decreased power of cardiac contraction.
Phosphorylation of troponin I by cyclic nucleotide-dependent protein kinases. As already mentioned, troponin I is phosphorylated by cAMP- and cGMP-dependent protein kinases. Under in vitro conditions, both protein kinases phosphorylate the same sites (Ser-23, -24) of cardiac troponin I [93, 94]. The cAMP-dependent protein kinase phosphorylates cardiac troponin I both in vitro and in vivo [4, 58]. Phosphorylation of troponin I by PKA results in a decrease in the sensitivity of the contractile apparatus to Ca2+. Therefore, after phosphorylation of troponin I the half-maximal ATPase activity and half-maximal tension are achieved at higher Ca2+ concentrations [95, 96]. This effect may be due to phosphorylation induced decreasing of affinity of troponin I to troponin C [97]. As a consequence of decreased interaction with troponin I, the regulatory sites of troponin C bind Ca2+ with lower affinity. Recently this suggestion was tested by gene engineering methods. It was shown that substitution of intact troponin I for a mutant protein with the replacement of Ser-23 and Ser-24 for Asp is accompanied by a decrease in Ca2+ sensitivity of the contractile apparatus [98]. In other words, introduction of a negative charge, whether it be by phosphorylation or by the replacement of Ser with Asp in positions 23 and 24 of troponin I, significantly decreases Ca2+ binding by troponin C. This effect may be caused by phosphorylation-induced changes in troponin I structure. Indeed, phosphorylation of Ser-23 and Ser-24 of a short troponin I peptide (residues 17-32) leads to large conformational changes [99]. Phosphorylation affects parameters of fluorescence of a label attached to the fifth residue of troponin I, i.e., it affects the structure in the vicinity of the site of phosphorylation [100]. Also, phosphorylation of Ser-23 and Ser-24 results in global changes of troponin I structure. Indeed, phosphorylation of intact troponin I by cAMP-dependent protein kinase is accompanied by a decrease in the distance between the fluorescent label fixed close to the N-termini of troponin I and a Trp residue located on the C-terminal end [101]. Phosphorylation also affects the fluorescence of labels attached in the central and C-terminal parts of troponin I, i.e., at the sites contacting the central alpha-helix and the N-terminal regulatory domain of troponin C [102]. Phosphorylation of troponin I in the complex with the N-terminal fragment of troponin C (residues 1-89) also results in a significant decrease in the affinity to Ca2+ [103]. Recently, a model was suggested for explaining the effect of troponin I phosphorylation on the Ca2+-binding properties of cardiac troponin C [104]. The data presented indicate that phosphorylation of Ser-23 and Ser-24 results in significant changes in troponin I structure and affects the interaction of troponin I with troponin C and the binding of Ca2+ by the regulatory sites of troponin C. Thus, phosphorylation of Ser-23 and Ser-24 of cardiac troponin I by protein kinases A and G decreases the Ca2+ sensitivity of the contractile apparatus.
Phosphorylation of troponin I and regulation of muscle contractile activity. Attempts to understand the relations between hormone action and changes in the contractile activity induced by troponin I phosphorylation have been undertaken for many years. It is well known that beta-adrenergic stimulation results in an increase in both the frequency and power of heart contraction [105]. Activation of beta1- or beta2-adrenoreceptors is accompanied by an increase in adenylate cyclase activity and therefore enhancing of protein kinase A activity. This leads to an increase in phosphorylation of troponin I, C-protein as well as of phospholamban (an activator of the Ca2+-ATPase of sarcoplasmic reticulum) [94, 106]. Stimulation of alpha1-adrenoreceptors in the presence of beta-receptor blockers is accompanied by an increase in phosphatidylinositol turnover and activation of protein kinase C. It is supposed that retardation of heart contraction induced by agonists of alpha1-adrenoreceptors may be connected with troponin I phosphorylation by protein kinase C [92]. Adenosine A1 receptors block adenylate cyclase. This may be the reason why stimulation of adenosine receptors leads to a decrease in phosphorylation of troponin I, C-protein, phospholamban, and phospholeman [107] as well as a decrease in the power and frequency of heart contraction. It is an oversimplification to explain all changes in heart contraction solely by phosphorylation of troponin I. Actually, the amplitude and frequency of heart contraction are determined both by the Ca2+ sensitivity of the contractile apparatus (i.e., depends on phosphorylation of troponin I and/or protein C) and by the fluctuation of Ca2+ in myoplasm (i.e., it depends on the functioning of the Ca2+-transporting membrane ATPases). The efficiency of the Ca2+-transporting systems may be regulated by phosphorylation of phospholamban and phospholeman [108]. Thus, the contractile activity of the heart depends on the coordinated phosphorylation of both regulatory proteins (troponin I and C-protein) and membrane proteins (phospholamban, phospholeman, and Ca2+ channels).
There is no doubt that phosphorylation of troponin I plays an important role in the regulation of the contractile activity of the heart. It is not surprising that different pathologies (such as heart failure, myocardial infarction, or arterial hypertension) affect the extent of troponin I phosphorylation [7, 29, 95]. Unfortunately, at present it is difficult to establish the relations between different forms of pathology and troponin I phosphorylation. However, there is no doubt that phosphorylation of troponin I by different protein kinases may significantly affect heart contractile activity.
III. TROPONIN T
Structure and Isoform Composition of Troponin T
Troponin T is a troponin component that interacts with tropomyosin and plays an important role in regulation of muscle contraction [56, 109]. The primary structure of troponin T from skeletal and cardiac muscle of several species of mammals, birds, and invertebrates have been described in the literature [110]. In most muscles troponin T is present in the form of multiple isoforms [111, 112]. The presence of several troponin T genes [110, 113] as well as alternative splicing [110, 114, 115] provide for the existence of the multiple troponin T isoforms. Troponin T genes contain several exons undergoing alternative splicing; thus, rat skeletal muscle theoretically could contain as many as 128 isoforms of troponin T differing in amino acid composition [116]. It is interesting that the scheme of alternative splicing of the primary RNA transcript is different for troponin T from different sources [117] and depends on the stage of ontogenesis.
Human cardiac muscle contains four troponin T isoforms, three of which are expressed in the fetus, and one isoform is characteristic for adult heart [115]. Reexpression of embryonic forms of troponin T both at the level of mRNA [115] and protein [115, 118] is observed during heart failure.
Charged residues are unevenly distributed in the troponin T structure. The N-terminal part including residues 1-59 is enriched in negatively charged residues, whereas the C-terminal part is enriched in positively charged residues. This distribution of charged residues results in a tendency of troponin T to aggregation at physiological salt concentration [56, 110].
Electron microscopy data indicate that in the isolated state or inside the thin filament (containing actin and tropomyosin), troponin T has the form of a comma or rod with a length of 185-205 Å. Troponin T is located in the groove of the actin helix and is extended along the filament [110, 119, 120].
Interactions of Troponin T with Other Components of the Thin Filament
Interaction of troponin T with tropomyosin. Greaser and Gergely [3] were the first to show that troponin T interacts with tropomyosin. Later this interaction was analyzed in detail. There are two tropomyosin-binding sites in the troponin T structure. One site is within residues 1-158 and the second is located close to the C-terminal end, within residues 159-259 [110, 121]. Interaction of the N-terminal chymotryptic peptide of troponin T (the so-called T1 fragment, residues 1-158) with tropomyosin is not dependent on Ca2+ [71]. A shorter fragment containing residues 71-151 also interacts with tropomyosin, although more weakly than the T1 fragment [109]. This finding indicates that residues 1-70 play an important role in the interaction with tropomyosin. Nevertheless, troponin T with deletion of the first 45 residues interacts with tropomyosin with affinity comparable to that of intact troponin T [122], and the short peptide containing residues 1-70 is unable to directly interact with tropomyosin [123]. The interaction of troponin T with tropomyosin is significantly reduced after removing residues 70-150 and is only slightly dependent on the presence of residues 1-69 or 151-158 [109]. Probably the main Ca2+-independent site of the troponin T--tropomyosin interaction is the site within residues 71-151. It is supposed that this alpha-helical region interacts with the alpha-helix of tropomyosin. Fragment T1 (residues 1-158 of troponin T) binds to the site located close to the C-terminus of the tropomyosin dimer [121] and overlaps the region of contacts of two tropomyosin dimers interacting head to tail.
The interaction of the troponin T site within residues 159-259 with tropomyosin depends on Ca2+ [71]. The troponin complex containing, instead of intact troponin T, the peptide containing residues 159-259 of troponin T interacts with tropomyosin with lower affinity but provides Ca2+-dependent regulation of muscle contraction as does the intact troponin complex [124]. The troponin T peptide consisting of residues 159-259 contains two tropomyosin-binding sites, both of which are located close to Cys-190 of tropomyosin. One of these tropomyosin-binding sites (residues 228-259 or residues 243-259) interacts only with tropomyosin, whereas the second site (residues 159-222 or 156-227) provides for the interaction of troponin T with both tropomyosin and troponin C [84, 121]. The troponin T site within residues 159-227 contains conservative heptads of hydrophobic residues [125]. Analogous heptad motifs are characteristic for tropomyosin structure. It is supposed that troponin T and a tropomyosin dimer form a triple coiled coil stabilized by hydrophobic interactions [110, 121]. It is worthwhile to mention that this site of troponin T can interact with troponin C and can be chemically cross-linked to Cys-190 of tropomyosin. The probability of cross-linking is increased in the presence of Ca2+ [84]. Thus, troponin T contains three tropomyosin-binding sites. The N-terminal site of troponin T (residues 1-159) provides for Ca2+-independent interaction with the C-terminus of one and the N-terminus of another tropomyosin dimer. The second site, lying within residues 156-227, can form a triple coiled coil with tropomyosin and contacts troponin C. And the third site, within residues 227 (243)-259 as well as the second site contacts tropomyosin close to its Cys-190. The interaction of the second and the third troponin T sites with tropomyosin is Ca2+-dependent.
Interaction of troponin T with troponin I. Under in vitro conditions troponin T binds troponin I with Ka ~ 8.5·106 M-1 [81]. Recently published data indicate that the troponin I-binding site is located in the C-terminal part of troponin T and contains residues 176-230 [84], residues 152-175 [126], or residues 202-259 [127] of troponin T. The site containing the C-terminal 100 residues of troponin T probably interacts with the troponin I site within residues 41-96. This interaction depends on the state of Cys-48 and Cys-64 of troponin I. Oxidation of these residues or their chemical modification decreases the interaction of troponin I and troponin T [84].
In the primary structure of troponin T there are two sites (residues 205-255 and 155-205) characterized by their tendency to form alpha-helices and containing hydrophobic heptad repeats. A similar property is characteristic for the site of troponin I lying within residues 53-106. It is supposed that troponin T interacts with its partners tropomyosin and troponin I by forming triple coiled-coil structures [51, 87].
Interaction of troponin T with troponin C. There is no consensus in the literature on the nature of the interaction between troponin T and troponin C. In early investigations chemical cross-linking of the whole troponin complex resulted in no cross-linking of troponin T and troponin C [128] or the probability of cross-linking of these troponin components was very low [86]. However, in the course of purification of cardiac troponin components, troponin C was copurified with troponin T [129, 130] and under certain conditions troponin C was bound to immobilized troponin T [55]. Troponin T also affects chemical modification of certain residues of troponin C [131]. The sites of troponin T within residues 159-259 [71] or 176-230 [132] seem to be responsible for an interaction with troponin C. The N-terminal globular domain and the central helix of troponin C (residues 1-100) play the main role in the interaction with troponin T [51]. This probably explains why troponin T affects chemical modification of Lys-37 located in the N-terminal part of troponin C [131].
Role of Troponin T in Regulation of Muscle Contraction
The asymmetric extended comma-shaped molecule of troponin T [120] provides contacts between troponin components, tropomyosin, and actin. The C-terminal globular domain of troponin T interacts with tropomyosin (in the vicinity of Cys-190), troponin components, and actin. The extended N-terminal part of troponin T forms the tail of the comma. This part of the molecule is highly variable due to the alternative splicing of short exons and interacts with the C-terminal end of one and the N-terminal end of another (adjacent) tropomyosin dimer [110]. In this way troponin T may affect the interaction of two neighboring tropomyosin molecules and tropomyosin polymerization [110] as well as the interaction of tropomyosin with actin [109]. It has been shown that a troponin complex containing mutant troponin T with deletion of the first 150 residues has much less influence on the interaction of tropomyosin with actin than the troponin complex containing intact troponin T [109]. As already mentioned, the N-terminal part of troponin T (residues 1-70) is characterized by high variability due to alternative splicing of short exons [116]. It is worthwhile mentioning that there is a strict correlation in synthesis of troponin T and tropomyosin isoforms in different muscle fibers. The isoforms synthesized in a given muscle fiber seem to match each other and provide for correct assembly of the thin filament [110].
Troponin T is important not only for fixing of the troponin components to the actin--tropomyosin filament. Troponin T plays an important role in the regulation of actomyosin ATPase activity. Addition of troponin T to the troponin I--troponin C complex provides for deeper inhibition of actomyosin ATPase in the absence of Ca2+ and to activation of actomyosin ATPase activity in the presence of Ca2+ [72]. It is supposed that the activating effect of troponin T is due to its direct interaction with troponin C inside the whole troponin complex [56].
The direct interaction of troponin T with actin is discussed in the literature [133]. The data of recent years indicate that the central part of troponin T (residues 68-126 of skeletal troponin T or residues 95-153 of cardiac troponin T), in addition to its interaction with troponin components, may directly contact actin [134]. In the absence of Ca2+, the troponin--tropomyosin complex is fixed on the actin filament through troponin I. In the presence of Ca2+, when the contacts of troponin I with actin become weak, troponin T plays a crucial role in fixing of the whole troponin complex on the actin filament [134].
Troponin T Phosphorylation
Troponin T can be phosphorylated by different protein kinases both in the isolated form and inside the whole troponin complex.
A special enzyme called troponin T kinase has been found in skeletal and cardiac muscles [135, 136]. This enzyme rapidly phosphorylates isolated troponin T and troponin T inside the whole troponin complex, transferring a phosphate group to Ser-1 [135, 136]. Troponin T kinase has been thoroughly characterized [135-137]. The enzyme belongs to the casein kinase II family [137]. It is important to mention that troponin T isolated from skeletal muscle contains about 0.8 mole of phosphate per mole of protein. The phosphate is predominantly bound to the Ser-1 of troponin T [110]. Therefore, it was suggested that troponin T kinase is involved in phosphorylation of troponin T in vivo. Unfortunately, the physiological role of troponin T phosphorylation remains enigmatic. Phosphorylation of troponin T by troponin T kinase does not affect the cation-binding properties of the troponin complex [138]. It is possible that phosphorylation of Ser-1 of troponin T somehow affects its interaction with tropomyosin or affects the turnover rate of troponin T.
Phosphorylase kinase also phosphorylates isolated troponin T and troponin T inside the whole troponin complex [110]. Highly purified preparations of phosphorylase kinase phosphorylate Ser-149--Ser-150 and/or Ser-156--Ser-157 and are unable to phosphorylate Ser-1 of skeletal troponin T [139].
Under in vitro conditions protein kinase C phosphorylates Thr-171 of isolated skeletal troponin T [140] as well as Thr-190, 199, and 200 of cardiac troponin T [141]. It is worthwhile to mention that Thr-171 of skeletal troponin T is homologous to Thr-199 of cardiac troponin T and that phosphorylation of this site is partially inhibited after incorporation of troponin T into the whole troponin complex [141]. This is an indirect proof of the fact that the C-terminal part of troponin T is involved in the interaction with other troponin components. Further investigations have shown that unfractionated preparations of protein kinase C predominantly phosphorylate Thr-280, i.e., the residue characteristic for only the cardiac isoform of troponin T [142]. Isoforms of protein kinase C differ in their ability to phosphorylate different sites of troponin T. The alpha-, beta-, and epsilon-isoforms of protein kinase C phosphorylate Thr-190, 199, and 280 and Ser-194 in bovine cardiac troponin T, whereas the zeta-isoform phosphorylates two still unidentified sites of troponin T [89]. Phosphorylation of troponin T by the alpha-isoform of protein kinase C results in a decrease in the maximal actomyosin ATPase activity and a decrease in its sensitivity to Ca2+ [89]. Phosphorylation of troponin T by the zeta-isoform of protein kinase C leads to a slight increase in the Ca2+ sensitivity of actomyosin ATPase and does not influence the maximal ATPase activity of actomyosin [89]. It is supposed that these effects are due to decreased affinity of phosphorylated troponin T to the actin--tropomyosin complex [143] or to phosphorylated troponin T-induced decrease in the rate of liberation of reaction products from the active site of myosin [144].
Treatment of cardiomyocytes with phorbol esters results in phosphorylation of troponin T [145]. Therefore, phosphorylation of troponin T by protein kinase C may affect the contractile activity of the heart by a hormone-modulated influence on protein kinase C activity [146].
IV. MODERN CONCEPTS OF THE REGULATION OF MUSCLE CONTRACTILE
ACTIVITY BY THE TROPONIN--TROPOMYOSIN COMPLEX
In the thin filament seven actin monomers interact with one tropomyosin dimer and one heterotrimeric troponin complex consisting of troponin C, troponin I, and troponin T [12, 120, 125]. This complex is designated as a regulatory unit. Since neighboring tropomyosins have so-called sticky ends they interact with each other, and a continuous thread of tropomyosin is located in the groove of the actin helix [12].
Sliding of the two types of filaments against each other form the molecular basis of muscle contraction. This sliding is induced by cycling interaction of myosin heads with actin. It is supposed that the dissociated myosin head hydrolyzes ATP, but the products of this reaction (ADP and inorganic phosphate) leave the active site of myosin very slowly. A myosin head containing ADP and phosphate interacts with certain sites on actin forming the so-called weak complex [147]. Afterwards this weak binding can be transformed into strong binding. Myosin heads moves over actin, inorganic phosphate is liberated from the active site of myosin, and the angle between the myosin head and thin filament is changed from 90 to 45°. This generates the power stroke and the actin filament slides against the myosin filament [147]. After liberation of ADP from the active site and binding of a new ATP molecule, the myosin head dissociates from actin and the cycle is repeated.
A priori one can suggest that troponin affects either the binding of the myosin head to actin or the transition between weak and strong binding of myosin to actin. Actually, the troponin--tropomyosin complex affects both these events. In the absence of Ca2+ troponin exerts double effects. Troponin I tightly interacts with certain sites of actin and fixes the whole troponin complex having dimensions comparable with 2-3 actin globules in such position that the myosin heads cannot even weakly interact with actin [148, 149]. Thus, troponin sterically blocks even the weak interaction of a part of the actin (about 50% of actin monomers) with myosin heads. In addition, troponin may affect the position of tropomyosin on the actin filament. In the absence of Ca2+, troponin increases the probability of movement of tropomyosin outside the actin groove. Tropomyosin occupies such a position that all other sites of actin are able to form only weak complexes with myosin heads but are unable to undergo the transition between the weak and strong binding of myosin [12, 149].
In the presence of Ca2+ the interaction of troponin I with actin is weakened. Troponin I and other troponin components interacting with troponin I are moving from actin to a larger distance [150] and the steric blocking of the actin--myosin interaction vanishes. Moreover, after saturation by Ca2+ troponin shifts the equilibrium between two states of tropomyosin on the actin filament towards the so-called turned-on state. In this state tropomyosin no longer interferes with either weak or strong binding of myosin with actin [148, 149].
The tropomyosin dimer has a rather rigid structure. Therefore, the troponin-induced movement of tropomyosin is propagated over the whole regulatory subunit containing seven monomers of actin [12, 125]. Shifting of tropomyosin from the “switched-off” to “switched-on” position is achieved not only under the influence of troponin. Binding of even one myosin head per seven actin monomers leads to the pushing of tropomyosin from the blocking position [12, 149]. This movement of tropomyosin is not directly connected with mechanical pushing of tropomyosin from one to another position. It is possible that binding of myosin induces conformational changes in the structure of actin and that these conformational changes are the primary reason for moving of tropomyosin into a new position [151]. It is worthwhile to mention that the “switching-on” of the regulatory subunit induced by the binding of the myosin head is accompanied by not only the transition of tropomyosin from one position to another, but also increases the Ca2+ affinity of troponin belonging to this regulatory subunit [149]. The actin filament is a highly cooperative system; therefore, the changes of the state of a given regulatory subunit affects the state of the neighboring regulatory subunits [152]. Strong binding of myosin with the given regulatory subunit increases the probability of strong binding of myosin with two neighboring regulatory subunits. Analogously, Ca2+ binding with troponin of the particular subunit increases the probability of movement of tropomyosin from the “switched-off” to the “switched-on” position. This movement occurs not only in this particular regulatory subunit, but also in the subunits located both to the left and to the right of the subunit saturated with Ca2+ [12, 152]. It is still unclear whether troponin affects only the number of myosin heads which are able to interact with actin [153] or the frequency of cyclic interaction of myosin heads with actin [152]. Anyhow, Ca2+-dependent movement of the troponin complex and/or tropomyosin in the grooves of actin are the key events in the mechanism of regulation. Let us try to imagine how Ca2+ binding may lead to the movement of the troponin--tropomyosin complex on the actin filament.
According to the recently published model of Tripet et al. [52], at low concentrations of Ca2+ troponin I interacts through its N-terminal peptide (residues 1-40) with the C-terminal domain of troponin C saturated with Mg2+ (Fig. 6a). Two inhibitory peptides of troponin I (the central inhibitory region, residues 96-115, and the C-terminal region, residues 140-148) interact with actin and fix the troponin--tropomyosin complex in the blocking state [52] (Fig. 6a). After initiation of contraction, Ca2+ binds to the regulatory sites located in the N-terminal domain of troponin C. This induces conformational changes in the N-terminal domain of troponin C, and it starts to interact with residues 116-131 of troponin I. The interaction of this site of troponin I with troponin C leads to dissociation of the C-terminal inhibitory peptide of troponin I from actin and to formation of a tight complex between the central inhibitory peptide of troponin I (residues 96-115) with the C-terminal domain of troponin C [52] (Fig. 6b). As a result of all these changes the interaction of troponin I with actin is weaken. Troponin together with tropomyosin moves from the blocking position deep into the groove of the actin filament.
The data presented show that the binding of Ca2+ by the N-terminal domain of troponin C leads to the conformational changes of its C-terminal domain. In the relaxed state, when the N-terminal domain is free of Ca2+, the C-terminal domain of troponin C interacts with residues 1-40 of troponin I (Fig. 6a). After binding of Ca2+ by the N-terminal domain of troponin C, its C-terminal domain stops to interact with residues 1-40 of troponin I and is switched to residues 96-115 of troponin I (Fig. 6b). The N-terminal site of troponin I (residues 1-96) does not exert inhibitory action on the ATPase activity of actomyosin [52, 154]. However, the N-terminal fragment of troponin I is important for troponin T-dependent activation of actomyosin ATPase in the presence of Ca2+ [31, 56, 72, 154]. As already mentioned, binding of Ca2+ by the regulatory site of troponin C leads to the movement of the N-terminal site of troponin I [155]. The troponin T-binding sites of troponin I are located in its N-terminal part (residues 40-96). Therefore, dissociation of the N-terminal site of troponin I from troponin C leads to the movement of the N-terminal site of troponin I towards troponin T and probably tropomyosin. This provides for a new location of the whole regulatory complex on the actin filament and optimizes the interaction of actin with myosin.Fig. 6. The model of Tripet et al. [52] illustrating the changes in the structure of the troponin--tropomyosin complex and describing the mechanism of regulation of striated muscle contraction. Actin is shown as grey spheres. Coiled-coil tropomyosin (Tm) is located in the groove of the actin filament. Troponin T (TnT) (wavy grey line) interacts with actin and tropomyosin. Troponin I (TnI) is shown as a strongly curved white line. In the relaxed state (a) the N-terminal site (residues 1-40) of troponin I interacts with the C-terminal globular domain of the dumbbell-like troponin C (TnC). The inhibitory sites of troponin I (residues 96-115 and 140-148) interact with actin. During contraction (b) the N-terminal domain of troponin C interacts with residues 116-131 of troponin I, forcing the inhibitory peptides of troponin I to dissociate from actin. The main inhibitory site (residues 96-115) starts to interact with the C-terminal domain of troponin C. The N- and C-terminal globular domains of troponin C are labeled by N and C, respectively.
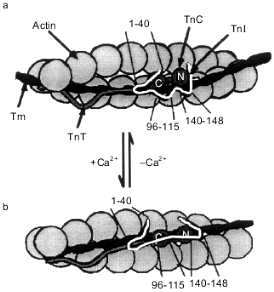
Thus, Ca2+-induced changes in the structure of the N-terminal domain of troponin C leads to the movement of certain sites of troponin I and to the weakening of the interaction of troponin I with actin (and probably with tropomyosin). Reorientation of certain sites of troponin I affects the position of troponin T on the actin filament. In addition, tropomyosin changes its position on the actin filament. All these events affect the accessibility of actin for interaction with myosin heads and as a result regulate muscle contraction.
This work was supported by the Russian Foundation for Basic Research (grant No. 98-04-48116).
REFERENCES
1. Ebashi, S., Kodama, A., and Ebashi, F. (1968)
J. Biochem., 64, 465-477.
2. Bailey, K. (1946) Nature, 157,
368-369.
3. Leavis, P. C., and Gergely, J. (1984) CRC Crit.
Rev. Biochem., 16, 235-305.
4. Perry, S. V. (1979) Biochem. Soc. Trans.,
7, 593-617.
5. Pan, B.-S., and Johnson, R. G. (1996) J. Biol.
Chem., 271, 817-823.
6. Wattanapermpool, J., Guo, X., and Solaro, R. J.
(1995) J. Mol. Cell. Cardiol., 27, 1383-1391.
7. Bodor, G. S., Anderson, P. A., Ladenson, J. H.,
Crimmins, D. L., Allen, P. D., and Oakeley, A. E. (1997)
Circulation, 96, 1495-1500.
8. Bhayana, V., and Henderson, A. R. (1995) Clin.
Biochem., 28, 1-29.
9. Hamm, C. W., and Katus, H. A. (1995) Curr.
Opin. Card., 10, 355-360.
10. Katrukha, A. G., Bereznikova, A. V., Esakova, T.
V., Pettersson, K., Lovgren, T., Severina, M. E., Pullki, K.,
Vuopio-Pullki, L. M., and Gusev, N. B. (1997) Clin. Chem.,
43, 1379-1385.
11. Kawasaki, H., and Kretsinger, R. H. (1994)
Protein Profile, 1, 343-390.
12. Tobacman, L. S. (1996) Annu. Rev.
Physiol., 58, 447-481.
13. Ikura, M. (1996) Trends Biochem. Sci.,
21, 14-17.
14. Babu, A., Su, H., Ryu, Y., and Gulati, J. (1992)
J. Biol. Chem., 267, 15469-15474.
15. Trigo-Gonzalez, G., Awang, G., Racher, K.,
Neden, K., and Borgford, T. (1993) Biochemistry, 32,
9826-9831.
16. Liu, W., Dotson, D. G., Lin, X., Mullen, J. J.,
Gouzalez-Garay, M. L., Lu, Q., and Putkey, J. A. (1994) FEBS
Lett., 347, 152-156.
17. Gagne, S. M., Li, M. X., and Sykes, B. D. (1997)
Biochemistry, 36, 4386-4392.
18. Satyshur, K. A., Rao, S. T., Pyzalska, D.,
Drendel, W., Greaser, M., and Sundaralingam, M. (1988) J. Biol.
Chem., 263, 1628-1647.
19. Herzberg, O., and James, M. N. G. (1988) J.
Mol. Biol., 203, 761-779.
20. Houdusse, A., Love, M. L., Dominguez, R.,
Grabarek, Z., and Cohen, C. (1997) Structure, 5,
1695-1711.
21. Slupsky, C. M., and Sykes, B. D. (1995)
Biochemistry, 34, 15953-15964.
22. Heidorn, D., and Trewhella, J. (1988)
Biochemistry, 27, 909-915.
23. Grabarek, Z., Tao, T., and Gergely, J. (1992)
J. Muscle Res. Cell Motil., 13, 383-393.
24. Potter, J. D., and Gergely, J. (1975) J.
Biol. Chem., 250, 4628-4633.
25. Gusev, N. B., and Barskaya, N. V. (1984)
Biochem. J., 220, 315-320.
26. Smith, L., Greenfield, N. J., and
Hitchcock-DeGregori, S. E. (1994) J. Biol. Chem., 269,
9857-9863.
27. Gulati, J., Akella, A. B., Su, H., Mehler, E.
L., and Weinstein, H. (1995) Biochemistry, 34,
7348-7355.
28. Li, M. X., Sykes, B. D., Smillie, L. B., Kay, C.
M., Tsuda, S., and Gagne, S. M. (1995) Biochemistry, 34,
8330-8340.
29. Li, P., Anversa, P., Meggs, L. G., Sonnenblick,
E. H., Cheng, W., Malhotra, A., and Hoffmann, P. A. (1997) Am. J.
Physiol., 272, H360-H370.
30. Szczesna, D., Guzman, G., Miller, T., Zhao, J.,
Farokhi, K., Ellemberger, H., and Potter, J. D. (1996) J. Biol.
Chem., 271, 8381-8386.
31. Malnic, B., Farah, C. S., and Reinach, F. C.
(1998) J. Biol. Chem., 273, 10594-10601.
32. Brito, R. M., Krudy, G. A., Negele, J. C.,
Putkey, J. A., and Rosevear, P. R. (1993) J. Biol. Chem.,
268, 20966-20973.
33. Sundaralingam, M., Drendel, W., and Greaser, M.
(1985) Proc. Natl. Acad. Sci. USA, 82, 7944-7947.
34. Babu, A., Gulati, J., Su, H., and Rao, V. G.
(1993) J. Biol. Chem., 268, 19232-19238.
35. Dobrowolski, Z., Xu, G.-Q., and
Hitchcock-DeGregori, S. E. (1991) J. Biol. Chem., 266,
5703-5710.
36. Ramakrishnan, S., and Hitchcock-DeGregori, S. E.
(1996) J. Biol. Chem., 35, 15515-15521.
37. Herzberg, O., and James, M. N. G. (1985)
Nature, 313, 653-659.
38. Herzberg, O., Moult, J., and James, M. N. G.
(1986) J. Biol. Chem., 61, 2638-2644.
39. Fujimori, K., Sorenson, M., Herzberg, O., Moult,
J., and Reinach, F. C. (1990) Nature, 345, 182-184.
40. Grabarek, Z., Tan, R.-Y., Wang, J., Tao, T., and
Gergely, J. (1990) Nature, 345, 132-135.
41. Da Silva, A. C. R., Dearanjo, A. H. B.,
Herzberg, O., Moult, J., Sorenson, M., and Reinach, F. C. (1993)
Eur. J. Biochem., 213, 599-604.
42. Sweeney, H. L., Brito, R. M., Rosevear, P. R.,
and Putkey, J. A. (1990) Proc. Natl. Acad. Sci. USA, 87,
9538-9542.
43. Gulati, J., Babu, A., and Su, H. (1992) J.
Biol. Chem., 267, 25073-25077.
44. Strynadka, N. C., James, M. N., Smillie, L. B.,
Li, M. X., Sielecki, A. R., and Cherney, M. (1997) J. Mol.
Biol., 273, 238-255.
45. Sia, S. K., Li, M. X., Spyracopoulos, L., Gagne,
S. M., Liu, W., Putkey, J. A., and Sykes, B. D. (1997) J. Biol.
Chem., 272, 18216-18221.
46. Spyracopoulos, L., Li, M. X., Sia, S. K., Gagne,
S. M., Chandra, M., Solaro, R. J., and Sykes, B. D. (1997)
Biochemistry, 36, 12138-12146.
47. Liao, R., Wang, C. K., and Cheung, H. C. (1994)
Biochemistry, 33, 12729-12734.
48. Krudy, G. A., Kleerekoper, Q., Guo, X., Howarth,
J. M., Solaro, R. J., and Rosevear, P. R. (1994) J. Biol. Chem.,
269, 23731-23735.
49. Olah, G. A., and Trewhella, J. (1994)
Biochemistry, 33, 12800-12806.
50. Vassylyev, D. G., Takeda, S., Wakatsuki, S.,
Maeda, K., and Maeda, Y. (1998) Proc. Natl. Acad. Sci. USA,
95, 4847-4852.
51. Farah, C. S., and Reinach, F. C. (1995) FASEB
J., 9, 755-767.
52. Tripet, B., van Eyk, J. E., and Hodges, R. S.
(1997) J. Mol. Biol., 271, 728-750.
53. Jha, P. K., Mao, C., and Sarkar, S. (1996)
Biochemistry, 35, 11026-11035.
54. Leszyk, J., Tao, T., Nuwaysir, L. M., and
Gergely, J. (1998) J. Muscle Res. Cell Motil., 19,
479-490.
55. Verin, A. D., and Gusev, N. B. (1988)
Biokhimiya, 53, 1235-1246.
56. Potter, J. D., Sheng, Z., Pan, B. S., and Zhao,
J. (1995) J. Biol. Chem., 270, 2557-2562.
56a. Kobayashi, T., Zhao, X., Wade, R., and
Collins, J. H. (1999) Biochemistry, 38, 5386-5391.
57. Dhoot, G. K., Gell, P. G. H., and Perry, S. V.
(1978) Exp. Cell. Res., 117, 357-370.
58. Perry, S. V. (1999) Mol. Cell. Biochem.,
190, 9-32.
59. MacGeoch, G., Barton, P. J., Vallins, P. J.,
Bhavzar, P., and Spurr, N. K. (1991) Hum. Genet., 88,
101-104.
60. Bhavzar, P. K., Brand, N. J., Yacoub, M. H., and
Barton, P. J. R. (1996) Genomics, 35, 11-23.
61. Schiaffino, G., Gorza, L., and Ausoni, S. (1993)
Trends Cardiovasc. Med., 3, 12-17.
62. Sasse, S., Brand, N. J., Kyprianou, P., Dhoot,
G. K., Wade, R., Arai, M., Periasamy, M., Yacoub, M., and Barton, P. J.
(1993) Circ. Res., 72, 932-938.
63. Syska, H., Wilkinson, J. M., Grand, R. J., and
Perry, S. V. (1976) Biochem. J., 153, 375-387.
64. Van Eyk, J. E., Strauss, J. D., Hodges, R. S.,
and Ruegg, J. C. (1993) FEBS Lett., 323, 223-228.
65. Levine, B. A., Moir, A. J. R., and Perry, S. V.
(1988) Eur. J. Biochem., 172, 389-397.
66. Pearlstone, J. R., Sykes, B. D., and Smillie, L.
B. (1997) Biochemistry, 36, 7601-7606.
67. Van Eyk, J. E., and Hodges, R. S. (1988) J.
Biol. Chem., 263, 1726-1732.
68. Talbot, J. A., and Hodges, R. S. (1981) J.
Biol. Chem., 256, 12374-12378.
69. Campbell, A. D., van Eyk, J. E., Hodges, R. S.,
and Sykes, B. D. (1992) Biochim. Biophys. Acta, 1160,
25-54.
70. Mak, A. S., Golosinska, K., and Smillie, L. B.
(1983) J. Biol. Chem., 258, 14330-14334.
71. Pearlstone, J. R., and Smillie, L. B. (1983)
J. Biol. Chem., 258, 2534-2542.
72. Farah, C. S., Miyamoto, C. A., Ramos, C. H. I.,
da Silva, A. C. R., Quaggio, R. B., Fujimori, K., Smillie, L. B., and
Reinach, F. C. (1994) J. Biol. Chem., 269, 5230-5240.
73. Pearlstone, J. R., and Smillie, L. B. (1995)
Biochemistry, 34, 6932-6940.
74. Ngai, S. M., Sonnichsen, H. M., and Hodges, R.
S. (1994) J. Biol. Chem., 269, 2165-2172.
75. Leszyk, J., Tao, T., Leavis, P. C., and Collins,
J. H. (1987) Biochemistry, 26, 7035-7042.
76. Van Eyk, J. E., Kay, C. M., and Hodges, R. S.
(1991) Biochemistry, 30, 9974-9981.
77. McKay, R. T., Tripet, B. F., Hodges, R. S., and
Sykes, B. D. (1997) J. Biol. Chem., 272, 28494-28500.
77a. McKay, R. T., Tripet, B. P., Pearlstone, J. R., Smillie, L. B., and
Syjes, B. D. (1999) Biochemistry, 38, 5478-5489.
78. Rarick, H. M., Tu, X.-H., Solaro, R. J., and
Martin, A. F. (1997) J. Biol. Chem., 272,
26887-26892.
79. Olah, G. A., Rokop, S. E., Wang, C. L.,
Blechner, S. L., and Trewhella, J. (1994) Biochemistry,
33, 8233-8239.
80. Stone, D. B., Timmins, P. A., Schneider, D. K.,
Krylova, I., Ramos, C. H. I., Reinach, F. C., and Mendelson, R. A.
(1998) J. Mol. Biol., 281, 689-704.
81. Ingraham, R. H., and Swenson, C. A. (1984) J.
Biol. Chem., 259, 9544-9548.
82. Chong, P. C. S., and Hodges, R. S. (1982) J.
Biol. Chem., 257, 9152-9160.
83. Sheng, Z., Pan, B.-S., Miller, T. E., and
Potter, J. D. (1992) J. Biol. Chem., 267,
25407-25413.
84. Chong, P. C. S., and Hodges, R. S. (1982) J.
Biol. Chem., 257, 11667-11672.
85. Hitchcock, S. E. (1975) Biochemistry,
14, 5162-5167.
86. Gusev, N. B., and Friedrich, P. (1980)
Biochim. Biophys. Acta, 626, 106-116.
87. Stefancsik, R., Jha, P. K., and Sarkar, S.
(1998) Proc. Natl. Acad. Sci. USA, 95, 957-962.
88. Noland, T. A., Jr., Kuo, J. F., Solaro, R. J.,
Averyhart-Fullard, V., Jideama, N. M., Raynor, R. L., and Guo, X.
(1995) J. Biol. Chem., 270, 25445-25454.
89. Jideama, N. M., Noland, T. A., Jr., Raynor, R.
L., Blobe, G. C., Fabbro, D., Kazanietz, M. G., Blumberg, P. M.,
Hannun, Y. A., and Kuo, J. F. (1996) J. Biol. Chem., 271,
23277-23283.
90. Noland, T. A., Jr., Kuo, J. F., Solaro, R. J.,
Blumberg, P. M., Kazanietz, M. G., Guo, X., Jideama, N. M., and Raynor,
R. L. (1996) Biochemistry, 35, 14923-14931.
91. Venema, R. C., and Kuo, J. F. (1993) J. Biol.
Chem., 268, 2705-2711.
92. Strang, K. T., and Moss, P. L. (1995) Circ.
Res., 77, 114-120.
93. Swiderek, K., Heilmeyer, L. M., Jr., Hoffmann,
F., Schachtele, C., Meyer, H. E., and Jaquet, K. (1990) Eur. J.
Biochem., 190, 575-582.
94. Sulakhe, P. V., and Vo, X. T. (1995) Mol.
Cell. Biochem., 149/150, 103-126.
95. McConnel, B. K., Bond, M., Norano, I., and
Moravec, C. S. (1997) Am. J. Physiol., 273,
H1440-H1451.
96. Malhotra, A., Buttrick, P., Bowman, J., and
Nakouzi, A. (1997) Mol. Cell. Biochem., 170, 99-107.
97. Al-Hillawi, E., Trayer, I. P., Trayer, H. R.,
and Bhandari, D. G. (1995) Eur. J. Biochem., 228,
962-970.
98. Dohet, C., Ruegg, J. C., Trayer, I. P., and
Al-Hillawi, E. (1995) FEBS Lett., 377, 131-134.
99. Quirk, P. G., Perry, S. V., Levine, B. A., Gao,
Y., and Patchell, V. B. (1995) FEBS Lett., 370,
175-178.
100.Dong, W. J., Cheung, H. C., Solaro, R. J.,
Xinng, J., and Chandra, M. (1997) Biochemistry, 36,
6745-6753.
101.Dong, W. J., Cheung, H. C., Solaro, R. J., She,
M., Xinng, J., and Chandra, M. (1997) Biochemistry, 36,
6754-6761.
102.Dong, W. J., Cheung, H. C., Gordon, A. M., and
Wang, C. K. (1997) Biophys. J., 72, 850-857.
103.Chandra, M., Dong, W.-J., Pan, B.-S., Cheung,
H. C., and Solaro, R. J. (1997) Biochemistry, 36,
13305-13311.
104.Jaquet, K., Lohmann, K., Czisch, M., Holak, T.,
Gulati, J., and Jaquet, R. (1998) J. Muscle Res. Cell Motil.,
19, 647-659.
105.Kaumann, A. J., Krause, E. G., Karczewski, P.,
Kuschel, M., Bartel, S., Lynham, J. A., and Sanders, L. (1996) Mol.
Cell. Biochem., 163/164, 113-123.
106.Kaumann, A. J., and Molenaar, P. (1997)
Naunyn Schmiedebergs Arch. Pharmacol., 355, 667-681.
107.Neumann, J., Watanabe, A. M., Scholz, H.,
Schmitz, W., Rask, H. T., Krause, E. G., Bartel, S., Bodor, G. S.,
Jones, L. R., and Gupta, R. C. (1995) J. Mol. Cell. Cardiol.,
27, 1655-1667.
108.Kranias, E. G., and Solaro, R. J. (1983)
Fed. Proc., 42, 33-38.
109.Fisher, D., Wang, G., and Tobacman, L. S.
(1995) J. Biol. Chem., 270, 25455-25460.
110.Perry, S. V. (1998) J. Muscle Res. Cell
Motil., 19, 575-602.
111.Pan, B. S., and Potter, J. D. (1992) J.
Biol. Chem., 267, 23052-23056.
112.Jin, J. P., Huang, Q. Q., Yeh, H. I., and Lin,
J. J. C. (1992) J. Mol. Biol., 227, 1269-1276.
113.Heywood, S. M., Thibailt, M. C., and Siegel, E.
(1983) Cell Muscle Motil., 3, 157-193.
114.Greig, A., Hirschberg, Y., Anderson, P. A. W.,
Hainsworth, C., and Malouf, N. N. (1994) Circ. Res., 74,
41-47.
115.Anderson, P. A. W., Greig, A., Mark, T. M.,
Malouf, N. N., Oakeley, A. E., Underleider, R. M., Allen, P. D., and
Kay, B. K. (1995) Circ. Res., 76, 681-686.
116.Breitbart, R. E., Nguyen, H. T., Medford, R.
M., Destree, A. T., Mahdavi, V., and Nadal-Ginard, B. (1985)
Cell, 41, 67-82.
117.Townsend, P. J., Barton P. J. R., Yacoub, M.
H., and Farza, H. (1995) J. Mol. Cell. Cardiol., 27,
2223-2236.
118.Solaro, R. J., Powers, F. M., Gao, L., and
Gwathmey, J. K. (1993) Circulation, 87, 38-43.
119.Ohtsuki, I., and Nagano, K. (1982) Adv.
Biophys., 15, 93-130.
120.Flicker, P. F., Phillips, G. N., Jr., and
Cohen, C. (1982) J. Mol. Biol., 162, 495-501.
121.Mak, A. S., and Smillie, L. B. (1981) J.
Mol. Biol., 149, 541-550.
122.Pan, B. S., Gordon, A. M., and Potter, J. D.
(1991) J. Biol. Chem., 266, 12432-12438.
123.Pearlstone, J. R., and Smillie, L. B. (1977)
Can. J. Biochem., 55, 1032-1038.
124.Schaertl, S., Lehrer, S. S., and Geeves, M. A.
(1995) Biochemistry, 34, 15890-15894.
125.Zot, A. S., and Potter, J. D. (1987) Ann.
Rev. Biophys. Biophys. Chem., 16, 535-559.
126.Pearlstone, J. R., and Smillie, L. B. (1978)
Can. J. Biochem., 56, 521-527.
127.Jha, P. K., Leavis, P. C., and Sarkar, S.
(1996) Biochemistry, 35, 16573-16580.
128.Sutoh, K., and Matsuzaki, F. (1980)
Biochemistry, 19, 3878-3882.
129.Gusev, N. B., Barskaya, N. V., Verin, A. D.,
Duzhenkova, I. V., Khuchua, Z. A. and Zheltova, A. O. (1983)
Biochem. J., 213, 123-129.
130.Katrukha, A. G., Bereznikova, A. V., Esakova,
T. V., Filatov, V. L., Bulargina, T. V., and Gusev, N. B. (1995)
Biochem. Mol. Biol. Int., 36, 195-202.
131.Hitchcock, S. E., Zimmerman, C. J., and
Smalley, C. (1981) J. Mol. Biol., 147, 125-151.
132.Leszyk, J., Collins, J. H., Leavis, P. C., and
Tao, T. (1988) Biochemistry, 27, 6983-6987.
133.Dahiya, R., Butters, C. A., and Tobacman, L. S.
(1994) J. Biol. Chem., 269, 29457-29461.
134.Hinkle, A., Goranson, A., Butters, C. A., and
Tobacman, L. S. (1999) J. Biol. Chem., 274,
7157-7164.
135.Gusev, N. B., Dobrovolskii, A. B., and Severin,
S. E. (1980) Biochem. J., 189, 219-226.
136.Villar-Palasi, C., and Kumon, A. (1981) J.
Biol. Chem., 256, 7409-7415.
137.Risnik, V. V., and Gusev, N. B. (1984)
Biochim. Biophys. Acta, 790, 108-116.
138.Dobrovol'skij, A. B., and Gusev, N. B. (1982)
Biochem. Int., 5, 473-480.
139.Risnik, V. V., Dobrovolskii, A. B., and Gusev,
N. B. (1980) Biochem. J., 191, 851-854.
140.Risnik, V. V., Vorotnikov, V. V., and Gusev, N.
B. (1987) Biomed. Biochim. Acta, 8/9, 444-447.
141.Noland, T. A., Jr., Raynor, R. L., and Kuo, J.
F. (1989) J. Biol. Chem., 264, 20778-20785.
142.Noland, T. A., Jr., and Kuo, J. F. (1993) J.
Mol. Cell. Cardiol., 25, 53-65.
143.Noland, T. A., Jr., and Kuo, J. F. (1992)
Biochem. J., 288, 123-129.
144.Mehegan, J. P., and Tobacman, L. S. (1991)
J. Biol. Chem., 266, 966-972.
145.Liu, J. D., Wood, J. G., Raynor, R. L., Wang,
Y. C., Noland, T. A., Jr., Ausari, A. A., and Kuo, J. F. (1989)
Biochem. Biophys. Res. Commun., 162, 1105-1110.
146.Gwathmey, J. K., and Hajjar, R. J. (1990)
Circ. Res., 67, 744-752.
147.Rayment, I., Holden, H. M., Whittaker, M.,
Yohn, C. B., Lorenz, M., Holmes, K. C., and Milligan, R. A. (1993)
Science, 261, 58-65.
148.Lehrer, S. S. (1994) J. Muscle Res. Cell
Motil., 15, 232-236.
149.Lehrer, S. S., and Geeves, M. A. (1998) J.
Mol. Biol., 277, 1081-1089.
150.Squire, J. M., and Morris, E. P. (1998)
FASEB J., 12, 761-771.
151.Cassell, M., and Tobacman, L. S. (1996) J.
Biol. Chem., 271, 12867-12872.
152.Butters, C. A., Tobacman, J. B., and Tobacman,
L. S. (1997) J. Biol. Chem., 272, 13196-13202.
153.Homsher, E., Kim, B., Bobkova, A., and
Tobacman, L. S. (1996) Biophys. J., 70, 1881-1892.
154.Van Eyk, J. E., Thomas, L. T., Tripet, B.,
Wiesner, R. J., Pearlstone, J. R., Farah, C. S., Reinach, F. C., and
Hodges, R. S. (1997) J. Biol. Chem., 272,
10529-10537.
155.Luo, Y., Wu, J.-L., Gergely, J., and Tao, T.
(1997) Biochemistry, 36, 11027-11035.
156.Sayle, R. A., and Milner-White, E. J. (1995)
Trends Biochem. Sci., 20, 374-376.