PCR Fragmentation of DNA
L. A. Zheleznaya1, V. G. Kossykh2, I. V. Svad'bina1, T. S. Oshman1, and N. I. Matvienko3*
1Institute of Theoretical and Experimental Biophysics, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 135-62192University of Texas, Medical Branch, Galveston, TX, USA
3Institute of Protein Research, Russian Academy of Sciences, Pushchino, Moscow Region, 142292 Russia; fax: (095) 924-0493; E-mail: nikmatv@vega.protres.ru
* To whom correspondence should be addressed.
Received September 23, 1998; Revision received January 20, 1999
A method has been developed to prepare random DNA fragments using PCR. First, two cycles are carried out at 16°C with the Klenow's fragment and oligonucleotides (random primers) with random 3'-sequences and the 5'-constant part containing the site for cloning with the site-specific endonuclease. The random primers can link to any DNA site, and random DNA fragments are formed during DNA synthesis. During the second cycle, after denaturation of the DNA and addition of the Klenow's fragment, the random primers can link to newly synthesized DNA strands, and after DNA synthesis single-stranded DNA fragments are produced which have a constant primer sequence at the 5'-end and a complementary to it sequence at the 3'-end. During the third cycle, the constant primer is added and double-stranded fragments with the constant primer sequences at both ends are formed during DNA synthesis. Incubation for 1 h at 37°C degrades the oligonucleotides used at the first stage due to endonuclease activity of the Klenow's fragment. Then routine PCR amplification is carried out using the constant primer. This method is more advantageous than hydrodynamic methods of DNA fragmentation widely used for "shotgun" cloning.
KEY WORDS: PCR amplification of DNA, Taq-polymerase, Klenow's fragment
One of the most widespread problems in obtaining recombinant DNA is fragmentation of donor DNA into random fragments. This arises in "shotgun" cloning when sequencing whole genomes and when gene fragment cloning using the technique of phage "displays".
Four methods for preparing random fragments are known: 1) hydrodynamic disruption of DNA by ultrasonic treatment [1], passage through capillaries under pressure [2], or dispersion of a DNA solution into fine drops [3]; 2) treatment of DNA with DNase I in the presence of manganese [4]; 3) treatment of DNA with endonuclease CviJI [5] under conditions when this site-specific endonuclease, yielding fragments with "blunt" ends on DNA cleavage, loses its specificity and cleaves DNA randomly; 4) T-PCR amplification [6]. The latter method for DNA amplification uses so-called tagged random primers with a random (randomized) sequence of nine or more nucleotides at the 3'-end and a constant marker sequence 15-20 nucleotides long at the 5'-end. PCR is done in two steps: first with the random primer which is hybridized in a random way with DNA at low temperature; then with an oligonucleotide identical to the constant part of the random primer. Thermostable DNA polymerase is used in both steps, and the primer is changed by gel-filtration of the DNA preparation synthesized at the first step of synthesis.
The first two methods are the most widely used [1, 2]. However, they have a significant drawback. The ends of the resulting fragments must be “polished”, i.e., treated with DNA polymerase (phage T4 or the Klenow's fragment) or with nuclease specific to single-stranded DNA. However, the efficiency of recombinant DNA formation even after "polishing" is lower by approximately one order of magnitude than with the use of site-specific endonucleases producing DNA fragments with "blunt" ends [7].
The use of endonuclease CviJI yielding random DNA fragments with "blunt" ends is complicated by its low availability.
By the first three methods, random DNA fragments with "blunt" ends are produced. Linkers, synthetic double-helical oligonucleotides with a necessary restriction site for cloning, are often ligated with the "blunt" ends because ligation of DNA fragments with blunt ends is less efficient than ligation of those with sticky ends. This is followed by treatment with the respective endonuclease, and only then the fragments are ligated with a vector molecule [3, 8]. Thus, the cloning of random fragments becomes a labor-consuming procedure. The fourth method was suggested only recently [6] and has been little studied for possible artifacts. The use of Taq-polymerase at the first step requires long regions of randomized sequence in the random primer and low annealing temperature at which Taq-polymerase functions very inefficiently. Furthermore, this step includes gel-filtration exchange of the primer, which greatly complicates the method.
We suggest here a new version of T-PCR amplification using the Klenow's fragment instead of Taq-polymerase in the first step. This increases the efficiency of DNA synthesis at this step and permits use of a random primer with a short randomized sequence; this must decrease the probability of artifact DNA synthesis. Moreover, in our method the random primer is removed by the endonuclease activity of the Klenow's fragment.
MATERIALS AND METHODS
The construction of plasmid pGEM-9Z(-)RII was described previously [9]. This plasmid contains an ~3600-bp fragment with the genes for EcoRII endonuclease and methylase integrated between XbaI and SalI sites in vector pGEM-9Z (Promega, USA). The greater part of the fragment, 2883 bp, includes EcoRII genes. The ends of the fragment, ~300 and ~430 bp, are not deciphered. The integrated fragment was cut out of the plasmid with endonucleases XbaI and SalI, purified by electrophoresis in agarose gel, and subjected to PCR fragmentation.
The Klenow's fragment of DNA polymerase I, the Stoffel's fragment of Taq-polymerase, phage T4 DNA ligase, and endonuclease BspMKI (an isoschizomer of SalI) were isolated in the Laboratory of Molecular Genetics, Institute of Protein Research. Polynucleotide kinase was a gift from Boehringer Mannheim (Germany). For DNA sequencing, we used [gamma-32P]ATP (Cluster Co., Obninsk, Russia).
The oligonucleotides, primers with the random 3'-end GCCGTCGACGAATTC9N and the constant primer GCCGTCGACGAATTC, were synthesized by M. G. Shlyapnikov (Institute of Biochemistry and Physiology of Microorganisms, Russian Academy of Sciences, Pushchino). The oligonucleotides were labeled and purified as described in [10].
DNA fragments were preparatively isolated using DEAE paper as described in [11]. Purification of plasmid and phage DNA, transformation of competent cells, and sequencing were performed as described in the manual by Maniatis, Fritsch, and Sambrook [12].
PCR fragmentation of DNA. The process of DNA fragmentation consisted of two steps. In the first step, first cycle, 50 µl of reaction mixture containing 1x PCR buffer (10 mM Tris-HCl, pH 8.3, 1.5 mM MgCl2, 50 mM KCl), 200 µM dNTP, and 0.05 µg DNA was heated for 5 min at 94°C, the mixture was transferred onto ice for 5 min, then kept for 5 min at 16°C, and 15 pmoles of random primer was added and incubated for 10 min at the same temperature. Three units of the Klenow's fragment were then added, and incubation was continued for another 10 min at 16°C. The process of heating, annealing, and DNA synthesis was then repeated once more, adding a new portion of the Klenow's fragment (second cycle). In the second cycle the random primer was not added. In the third cycle 15 pmoles of the constant primer together with the Klenow's fragment was introduced, and the procedure of DNA synthesis was carried out at 37°C for 1 h followed by DNA denaturation at 94°C for 5 min and cooling at 0°C.
In the second step, 40 pmoles of the constant primer was added to the mixture and 25 cycles of PCR amplification were run with Stoffel's fragment of Taq-polymerase in regimes of 30 sec at 94°C, 30 sec at 50°C, and 1 min at 70°C.
As controls we used reaction mixtures where one of the following components was absent: DNA, Klenow's fragment, random primer, or constant primer.
RESULTS AND DISCUSSION
The idea of PCR fragmentation of DNA originates from the method of preparing highly radioactive DNA probes with random hexameric oligonucleotides used as primers in the DNA polymerase reaction [13-15]. Random DNA fragments complementary to various regions of the DNA template are formed in the process of polymerization. It would be tempting to use such a technique for PCR amplification, and in this case a sufficient number of random fragments could be obtained for cloning even with small amounts of initial DNA. Moreover, a site for endonuclease could be introduced into the fragment ends, which would facilitate their subsequent integration into the vector molecules. However, short primer oligonucleotides with a length of few nucleotides cannot hybridize with DNA at functioning temperatures of thermostable DNA polymerases.
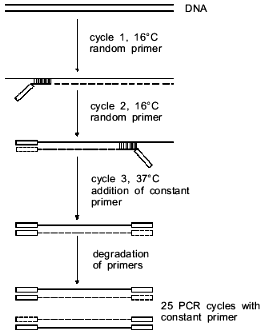
To overcome this circumstance, PCR amplification was carried out in two steps (Fig. 1). In the first step the DNA polymerase I Klenow's fragment was used. In this step in the two first cycles we used the oligonucleotide (random primer) with the randomized nucleotide sequence at the 3'-end and the constant sequence at the 5'-end with the site used for cloning endonuclease. Hybridization of the oligonucleotide and DNA synthesis was done at 16°C. Here the random primer molecules anneal to any DNA regions with their 3'-end, and random fragments appear as a result of synthesis. After repeated DNA denaturation and addition of the Klenow's fragment at the second cycle, the random primer anneals to the newly synthesized DNA strands, and single-stranded DNA fragments are formed after DNA synthesis with a nucleotide constant sequence at the 5'-end and a complementary to it sequence at the 3'-end. In the third cycle a constant primer is added and double-stranded fragments are formed with a constant primer sequence at both ends of the fragment during DNA synthesis. Subsequent prolonged incubation at 37°C cleaves unhybridized primers with the Klenow's fragment, in particular, of the random primer which in subsequent cycles of PCR amplification with Taq-polymerase could result in artifact DNA synthesis.
In the second stage, the DNA fragments with the constant primer at the ends are amplified in a routine PCR reaction with Taq-polymerase when one constant oligonucleotide is used as the primer.Fig. 1. General scheme for PCR amplification of DNA. Thin line, initial DNA; dashed line, DNA synthesized in the given cycle; thick line, DNA synthesized in the previous cycle. Rectangles denote the sequence of the constant primer or that complementary to it. Vertical lines show the region of complementarity of randomized sequence of the random primer and DNA.
In the present study two DNA polymerases, from E. coli and from Thermus aquaticus, function consecutively (in our case the Taq DNA polymerase Stoffel's fragment). The two consecutive steps should be done without a change of the buffer. So, we tested the efficiency of the Klenow's fragment in a standard buffer (10 mM Tris-HCl, pH 7.5, 10 mM MgCl2) and in a PCR buffer (10 mM Tris-HCl, pH 8.3, 50 mM KCl, 1.5 mM MgCl2). To this end, the “universal” primer, a heptadecameric oligonucleotide used for sequencing of M13 recombinants, was hybridized with single-stranded DNA of phage M13mp18, and synthesis of DNA was done in the presence of all four deoxynucleoside triphosphates, with the dATP being labeled. After synthesis the percentage of acid-insoluble label was measured by precipitation on GFC filters. The results of three independent experiments (data not given) showed that the Klenow's fragment functions with approximately the same efficiency both in the buffer for PCR and in the standard buffer, and, consequently, both steps can be carried out without a change of the buffer.
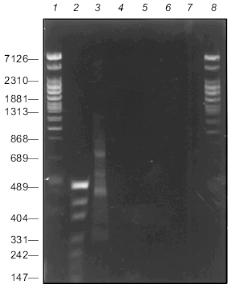
Figure 2 presents the results of PCR fragmentation of a 3.6-kb DNA fragment and corresponding controls. It is seen that the products of synthesis are present only in the mixture containing all the components of the reaction, while in the control mixtures DNA synthesis is not observed. These results indicate that the first step with the use of the random primer is absolutely necessary for PCR amplification. During the synthesis a heterogenous in length spectrum is formed of fragments with lengths of 100 to 1400 bp. Here, weak discrete bands are seen against a smeared background. They evidently appear as a result of highly effective hybridization of random primer with sequences enriched with GC pairs. Moreover, the source of such bands might be the degenerated homology of the constant primer sequence and that of the amplified DNA region (see below, discussion of the structure of the integrated fragment left end in clone 3, table).
Characteristics of sequenced clones containing gene fragments of the EcoRII systemFig. 2. Electrophoresis of products of PCR fragmentation in 1.5% agarose gel: 1, 2, 8) fragment length markers; 1, 8) phage T7 DNA cleaved with Bli736; 2) plasmid pUC DNA cleaved with HpaII. Lengths of some fragments in bp are given on the left; 3) complete reaction mixture; 4, 5, 6, 7) one of the components is absent: 4) DNA; 5) random primer; 6) Klenow's fragment; 7) constant primer.
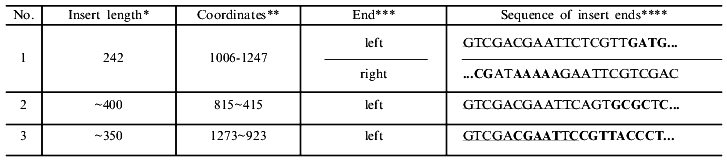
*Length of first the clone determined precisely by sequencing data, that of the second and third approximately by cleavage of recombinant DNAs with endonuclease BspMKI, so coordinates of the right end are given with the sign ~.
**Nucleotide sequence of genes of the EcoRII system obtained after merging of gene sequences of methylase [16] and endonuclease [17, 18]. Nucleotide 1 corresponds to the first nucleotide of the fragment sequenced in [16].
***The left end of the insert is adjacent to the region of the universal primer.
****Part of the constant primer sequence is underlined; bold letters denote nucleotide sequences of the genes corresponding to the regions. Ordinary letters within the defined regions correspond to mismatches (see text).
A large part of DNA synthesis can be artificial and connected with primer multimerization both during preparation of highly radioactive probes using the random primer method [15] and during T-PCR [6]. To be convinced that the resulting DNA is not an artifact of synthesis and represents random fragments of the DNA used for PCR, the PCR products were treated with endonuclease BspMKI, and fragments with lengths of 100 to 600 nucleotides were purified by electrophoresis in 1.5% agarose gel using DEAE paper. After elution, the fragments were desalted by dialysis and integrated into vector M13mp18. Three random clones producing colorless plaques were sequenced. The table presents data on these clones. The insert of one clone was completely sequenced, while the size of the fragments in the second and third clones exceeded that of the unambiguous region, so the sequence of only one end of the insert is given. From analysis of the end sequences of the inserts, a conclusion can be drawn on how the respective fragments appeared and how their integration into the vector took place.
In the case of clone 1, the left end appeared as the result of complementarity of four GATG nucleotides at the 3'-end of the random primer with the sequence in the vicinity of nucleotide 1006 of the methylase gene. The right end of this insert appeared as a result of complementarity of nine nucleotides with two mismatches of the 3'-end of the randomized primer 3'-GCTATTTTT-5' with the methylase gene sequence in the vicinity of nucleotide 1247 of 5'-CGCCAAAAA-3'. The left end of clone 2 originates as a result of complementarity of six nucleotides of primer 3'-CTCGCG-5' with the methylase gene sequence in the vicinity of nucleotide 815 of 5'-GCGCGC-3' with one mismatched C--T. The resulting data is in good accord with that published in [19], where it was shown that complementary pairing of two neighboring 3'-end primer nucleotides is sufficient for priming DNA synthesis with the use of the Klenow's fragment, and that at a greater length of the complementary region mismatched bases within the region do not hinder initiation of DNA synthesis. It follows also that the length of the randomized region at the 3'-end of the random primer can be decreased to 4-6 nucleotides, which facilitates reduction of artifact DNA synthesis [6, 15].
The left end of clone 3 most likely did not appear as the result of hybridization of the primer random region with DNA, which seems highly improbable due to the long length of the complementary region, but stems from hybridization of the constant primer with formation of a heteroduplex of 7 nucleotides of the constant primer, 5'-CGAATTC-3', and of 6 nucleotides of the methylase gene in the vicinity of nucleotide 1273, 3'-GCTTAG-5'. These events are probably one of the reasons for the appearance of discrete bands in the spectrum of the resulting fragments.
It should be noted than the use of one primer during the PCR hinders the formation of primer dimers, as was shown with the use of the so-called HANDS system [20]. The dimers resulting from random hybridization of the 3'-ends form a hairpin (Fig. 3a) after denaturation and annealing and so cannot effectively hybridize with the primer and undergo amplification.
Though such hairpin structures cannot be amplified, they can hinder the cloning of amplified products if the primer contains a site for the endonuclease used during cloning. Upon cleavage of the hairpin with endonuclease, in our case with BspMKI, a sticky end is formed which can attach itself to the sticky end of the linearized vector DNA and deactivate it. To avoid this, we purified the fragments by electrophoresis, though in many other aspects of the application of the suggested method this procedure may be not required, e.g., on preparation of highly radioactive probes for hybridization.Fig. 3. DNA structure formed during PCR amplification: a) formation of random primer dimer and hairpin structure; b) branching DNA structure formed from PCR amplification products after annealing. Rectangles at the strand ends denote the sequence of the constant primer or the sequence complementary to it.
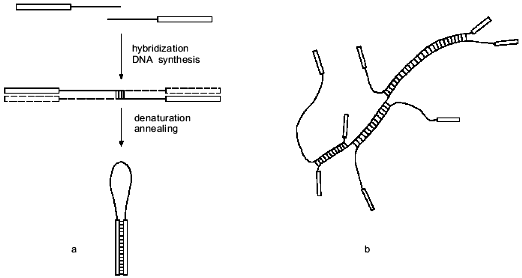
It is also noteworthy that in the suggested method intact DNA polymerase I and Taq-polymerase cannot be used instead of the Klenow's and Stoffel's fragments. Branching structures must form in the process of annealing during amplification (Fig. 3b); these would be cleaved by intact enzymes when they approach the points of branching [21, 22], while the Klenow's and Stoffel's fragments resolve the branching structures by displacing the strand [22, 23].
In conclusion, it should be noted that the suggested method of obtaining random fragments with the use of two polymerases has some advantages over the earlier described T-PCR method with decreased annealing temperature at the first cycles of PCR amplification, when Taq-polymerase is used in both steps [6]. First, there is no necessity to purity the mixture from the random primer by gel-filtration. Prolonged incubation at 37°C after the third cycle must result in degradation of the primers used in the first step with the Klenow's fragment. Second, with the suggested method quite short randomized sequences with lengths of 4-6 nucleotides at the 3'-end of the random primer can be used; this must decrease the probability of formation of dimers and primer multimers.
The proposed method can also be successfully used to obtain highly labeled radioactive DNA probes, which are available in very limited amounts. Artifact DNA synthesis is reduced to a minimum since one primer is used at the second step for amplification.
This study was supported by CRDF grant RB1-188.
REFERENCES
1.Dienenger, P. (1983) Anal. Biochem.,
129, 216-223.
2.Schriefer, L. A., Gebauer, B. K., Qui, L. Q. Q.,
Watersonn, R. H., and Wilson, K. (1990) Nucleic Acids Res.,
18, 7455.
3.Anderson, B., Wentland, M. A., Ricafrente, J. Y.,
Liu, W., and Gibbs, R. A. (1996) Anal. Biochem., 236,
107-113.
4.Anderson, S. (1981) Nucleic Acids Res.,
9, 3015-3027.
5.Fitzgerald, M. C., Skowron, P., van Etten, J. L.,
Smith, L. M., and Mead, D. A. (1992) Nucleic Acids Res.,
20, 3753-3762.
6.Grothues, D., Cantor, C. R., and Smith, C. L.
(1993) Nucleic Acids Res., 21, 1321-1322.
7.Burland, V., Daniels, D. L., Plunkett, G., III, and
Blattner, F. R. (1993) Nucleic Acids Res., 21,
3385-3390.
8.Povinelli, C. M., and Gibbs, R. A. (1993) Anal.
Biochem., 210, 16-26.
9.Matvienko, N. N., Zheleznaya, L. A., Chernysheva,
E. E., Bur'yanov, Ya. I., and Matvienko, N. I. (1997) Biochemistry
(Moscow), 62, 1314-1318 (Russ.).
10.Matvienko, N. N. Kramarov, V. M., Zheleznaya, L.
A., and Matvienko, N. I. (1993) Biochemistry (Moscow),
58, 1137-1151 (Russ.).
11.Winberg, G., and Hammarskjold, M.-L. (1980)
Nucleic Acids Res., 8, 253-264.
12.Sambrook, J., Fritsch, E. F., and Maniatis, T.
(1989) Molecular Cloning: A Laboratory Manual, Cold Spring
Harbor Laboratory Press, Plainview, New York.
13.Fienberg, A. P., and Vogelstain, B. (1983)
Anal. Biochem., 132, 6-13.
14.Fienberg, A. P., and Vogelstain, B. (1984)
Anal. Biochem., 137, 266-267.
15.Suganuma, A., and Gupta, K. C. (1995) Anal.
Biochem., 224, 605-608.
16.Som, S., Bhagwat, A. S., and Friedman, S. (1987)
Nucleic Acids Res., 15, 313-331.
17.Bhagwat, A. S., Johnsom, B., Weule, K., and
Roberts, R. J. (1990) J. Biol. Chem., 265, 767-773.
18.Kossykh, V. G., Repik, A. V., Kaliman, A. V.,
Bur'yanov, Ya. I., and Baev, A. A. (1989) Dokl. AN SSSR,
308, 1497-1499.
19.King, J. C., Fairley, C. F., and Morgan, W. F.
(1996) J. Biol. Chem., 271, 20450-20457.
20.Brownie, J., Shawcross, S., Theaker, J.,
Whitcombe, D., Ferrie, R., Newton, C., and Little, S. (1997) Nucleic
Acids Res., 25, 3235-3241.
21.Lyamichev, V., Brow, M. A. D., and Dahlberg, J.
E. (1993) Science, 260, 778-783.
22.Tombline, G., Bellizzi, D., and Sgaramella, V.
(1996) Proc. Natl. Acad. Sci. USA, 93, 2724-2728.
23.Walker, G. T. (1993) PCR Methods Applic.,
3, 1-6.