Affinity Purification of Alu-DNA-Repeat-Binding Proteins from Human Somatic Cells
D. V. Luk'yanov*, G. F. Reshetnikova, and O. I. Podgornaya
Institute of Cytology, Russian Academy of Sciences, Tikhoretskii pr. 4, St. Petersburg, 194064 Russia; fax: (812) 520-9703; E-mail: podg@ivm.stud.pu.ru* To whom correspondence should be addressed.
Received June 22, 1998; Revision received August 28, 1998
A 66-kD Alu-DNA-repeat binding protein was identified in human somatic cell nucleoplasm. Gel shift assay, southwestern blotting, and affinity purification on DNA attached to a carrier were used. A 60-kD protein copurified with the 66-kD protein during affinity purification, probably due to protein--protein interactions. The gel shift assay reveals multiple complexes with exponential dependence of their relative mobility. The short binding site of the 66-kD protein was defined with the help of synthetic oligonucleotides. It is localized between the A and B boxes of RNA polymerase III promotor and is the same as that reported for the Alu-binding protein from human spermatozoids. The same short binding site, the similarity of the isolation procedure from germ and somatic cells, and similar binding properties and molecular masses suggest homology of the two proteins. The relationship of the proteins we studied and the Alu-DNA-binding proteins described in the literature is discussed.
KEY WORDS: site-specific DNA--protein binding, Alu-repeat, Alu-DNA-binding proteins
Short interspersed repeated DNA elements (SINEs) are found in genomes of primates, rodents, and other higher eukaryotes [1]. Alu repeats are the largest class among these elements in the human genome. They are about 300 bp long and comprise about 3-6% of the haploid human genome. Alu repeats are flanked by direct repeats, have a dimer-like structure, and contain RNA polymerase III promoter which consists of A and B boxes in the left monomer [2]. There is a 10-20 bp A-rich region at the 3´ end. These features characterize Alu repeats as retroposons [3]. There are old and young subfamilies among Alu family repeats [4]. The concept that retroposons constitute "selfish DNA" begins to seem more and more impossible, and this drives an active search for the function of SINEs. Some hypotheses about the possible functions of Alu repeats in a cell are based on indirect experimental support as listed below.
1. Alu repeats are mobile regulators of transcription which are able to switch on or off certain genes [5-7].
2. The sequence between the A and B boxes in Alu reveals homology with the origins of DNA replication of some viruses [8]. It has been suggested that Alu repeats can be additional origins of DNA replication (subori) and lead the replication fork [9].
3. It has been shown that R bands in chromosomes where actively transcribing genes are situated are enriched with Alu repeats [10]. The mechanisms of origin and maintenance of structure of active chromatin in the nucleus are unknown. It is possible that Alu repeats may take part in it.
4. Alu repeats are actively transcribed by RNA polymerase III in vitro while being transcriptionally inactive in vivo [11]. The mechanisms of this inactivation are still not known. There is a scarce amount of Alu-RNA in human cells [12]. It was suggested that they are transcribed from a small Alu subfamily by RNA polymerase III. So, special mechanisms of transcription regulation must exist in cells providing selection of this subfamily from inactive Alu family repeats. It is possible that this selected transcription is directed by sequences of DNA around Alu repeats rather than their own features [13]. Transcripts from Alu repeats form stable Alu--RNP particles. Its functions remain unknown. It was suggested that they take part in regulation of protein translation [12].
Specific DNA-binding proteins are necessary for performance of any function of the Alu repeats mentioned above. So, identification and characterization of Alu-binding proteins may help to understand the functional role of Alu repeats in a cell.
The existence of Alu-repeat-binding proteins was proved a long time ago. Such proteins have been found in different cells by different research groups. In HeLa cells, two proteins of approximately 80 and 40 kD were described by Perelygina et al. [14]. An 80-kD protein was found by Chesnokov et al. [15] using southwestern blotting. Proteins of 120 and 35 kD from HeLa cells were described by Chiang and Vishwanatha [16]. A 60-kD protein from human sperm was reported [17]. It was shown that these proteins bind either the DNA sequence between the A and B boxes [18-21] or the B box and site downstream from it [22, 23] (see Fig. 4). But the characterization of all of these proteins is not exhaustive, and the relations between the proteins reported by different groups are not known.
In this study we revealed and defined the recognition site of the 66-kD Alu-binding protein from nucleoplasm of human somatic cells. The coincidence of binding sites and similarity of molecular masses and binding characteristics suggest that the revealed protein is homologous to the 60-kD protein from human spermatozoids [17].
MATERIALS AND METHODS
Isolation of nuclei and nuclear extract. Nuclei from a placenta or a human embryonal liver were isolated according to methods described [24]. All solutions also contained 1 mM phenylmethylsulfonyl fluoride (PMSF). Nuclear extract was obtained by addition of an equal volume of buffer containing 10 mM Tris-HCl, pH 8.0, 0.1 mM MgCl2, 0.2 mM PMSF, 0.5 mM dithiothreitol (DTT), 5% glycerol, and 0.35 M NaCl. After incubation for 1 h at 4°C the solution was centrifuged at 5000g for 10 min and the supernatant was used for subsequent work.
DNA, plasmids, and oligonucleotides. Plasmid pAL was prepared by recloning the Alu fragment from plasmid Blur8 [25] into plasmid pUC19 [14]. Ultrasonicated E. coli genome DNA or linearized pUC19 were used as nonspecific competitor in the gel mobility shift assay (GMSA). The plasmid was purified according to a conventional method and additionally purified in a CsCl gradient [26]. Complementary oligonucleotides 5´-GATCTGTAATCCCA-3´ and 5´-GATCTGGGATTACA-3´ were synthesized by SibEnzim (Novosibirsk, Russia). They were annealed in equimolar amount at 60°C for 1 h and incubated with nonlabeled or labeled dNTP and Klenow's fragment of DNA polymerase I from E. coli and purified by electrophoresis in 6% polyacrylamide gel [26]. The 270-bp length Alu fragment was excised from plasmid pAL using BamH I, labeled with [alpha-32P]dATP using a Klenow's fragment, and purified by electrophoresis in 0.5% agarose gel [26]. Subfragments of the Alu fragment were obtained by digestion of the labeled Alu with Hae III and separated by electrophoresis in 6% polyacrylamide gel [26].
Ion-exchange chromatography on DEAE-Sepharose. A column of DEAE-Sepharose (30 × 1.5 cm) was equilibrated with buffer containing 15 mM Tris-HCl, pH 8.0, 0.1 mM EDTA, 0.5 mM DTT, 0.1 mM PMSF, 5% glycerol, and 50 mM NaCl. Before application onto the column, 7 ml of nuclear extract (protein concentration 15 mg/ml) was diluted sevenfold with the equilibrating buffer. Proteins were eluted with 40 ml of a 0.05-1 M linear gradient of NaCl in the same buffer. All procedures were carried out at room temperature. Fractions of 1 ml were collected and stored at -20°C.
Gel mobility shift assay. Specific DNA-binding activity was revealed by the gel mobility shift assay (GMSA) [27]. The incubation mixture (20-70 µl) usually contained 5 ng of labeled Alu fragment or 0.5 ng of labeled oligonucleotide, 2-50 µl (0.1-10 µg protein) of DEAE fraction or affinity purified proteins and 0.1-7.0 µg of competitive DNA in retardation buffer containing 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1 mM DTT, and 5% glycerol. The mixture was incubated for 30 min at room temperature. Electrophoresis (40 mA, 3 h) was performed in 3.5% polyacrylamide gels in 0.5× TAE buffer (40 mM Tris-acetate, 2 mM EDTA) [26]. Gels were dried and exposed to Kodak XAR-5 films (USA).
Southwestern blotting. Proteins were resolved in a 7.5% gel by SDS-PAGE [28] and electrotransferred onto a nitrocellulose membrane [29]. Membrane filters were incubated in TBS buffer (15 mM Tris-HCl, pH 7.5, 150 mM NaCl) containing 0.5% dry milk overnight at 4°C. Then these filters were incubated in the retardation buffer with labeled Alu fragment (30 ng/ml). The mixture contained also 0.1% bovine serum albumin and nonspecific competitor DNA with concentration up to 12 µg/ml. Following three 5-min washings using the same buffer without DNA, the filters were exposed to Kodak XAR-5 films.
Affinity chromatography. Plasmid pAL was treated with EcoR I and Hind III. The Alu fragment obtained was purified by electrophoresis in a 0.5% agarose gel. The fragment (20 µg) was attached to cellulose (200 µl) using 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride as described [30]. DEAE fraction containing Alu-binding activity was incubated with immobilized DNA in an Eppendorf tube for 30 min at room temperature. The incubation mixture (300 µl) contained 150 µl of DEAE fraction and excess nonspecific competitor DNA in the retardation buffer. Following four 5-min washings using 10 volumes of the retardation buffer, the bound proteins were eluted with 1 M NaCl in the same buffer (200 µl) and analyzed by GMSA and SDS-PAGE [28].
RESULTS
Proteins with specificity in binding to Alu repeats were partially purified from nuclear extract of placenta cells according to procedure described in [14] except that proteins were eluted from DEAE column by a linear gradient of NaCl. The gel mobility shift assay (GMSA) was used to test the resulting fractions for Alu-binding activity. The peak of activity came eluted at about 0.25 M NaCl in 3 ml. Fractions with maximal activity were pooled together and used in the following work.
Figure 1a shows the result of the GMSA applied to active fractions. The intensity of the faster DNA--protein complexes decreased as the intensity of slower ones increased up to some amount of nonspecific competitive DNA (Fig. 1a, lanes 2-6). The relative mobility of the complexes has exponential dependence, i.e., the difference in the mass of the complexes is the same. This could be due to protein multimerization during formation of complexes. The protein in complexes may be a heterodimer but different complexes are not a result of the binding of different proteins.
The same procedure of ion-exchange chromatography of Alu-binding proteins was applied to the nucleoplasm of human embryonal liver cells. Very close similarity of Alu--protein complexes and their mobility in the GMSA test in comparison with placenta cell nucleoplasm was observed (data not shown).Fig. 1. Gel mobility shift assay (GMSA) of Alu fragment with DEAE-fraction (a) and affinity purified proteins (b). a) F, free fragment; 1-8) 10 µl protein (approximately 10 µg total protein) added in the presence of 50-, 75-, 100-, 150-, 300-, 450-, 1500-, and 7000-fold excess of the competitive DNA, respectively (the triangle below shows the increase of competitor). b: 1) conditions as in (a), lane 4; 2-4) mixture contains proteins obtained from different affinity columns in the presence of increasing amount of competitive DNA (200-, 500-, and 1000-fold excess of the competitive DNA during chromatography, respectively). The competitive DNA is absent from the retardation mixture. Arrows at the left and at the right mark the DNA--protein complexes.
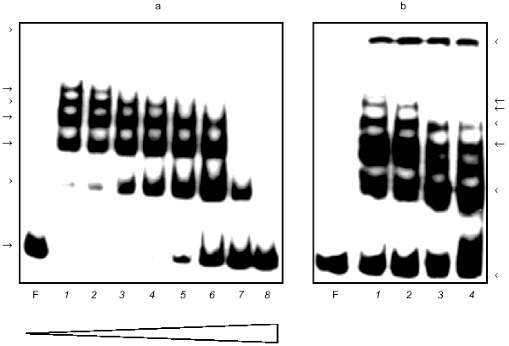
Affinity chromatography was used for further purification of Alu-binding proteins. DEAE active fraction was incubated with Alu fragments immobilized to the cellulose in the presence of different amounts of nonspecific competitive DNA. Proteins eluted from the column were tested by the GMSA (Fig. 1b). The concentration of NaCl in the incubation mixture did not exceed 0.25 M. It was adjusted by dilution of the eluate. The same volume of eluates obtained after chromatography in the presence of increasing amounts of competitive DNA is loaded on different lines. All fractions eluted from the column have Alu-binding activity which decreased insignificantly while amounts of competitor were increased (Fig. 1b, lanes 2-4). The complexes formed have the same appearance as in the case of the DEAE fraction (Fig. 1, a and b).
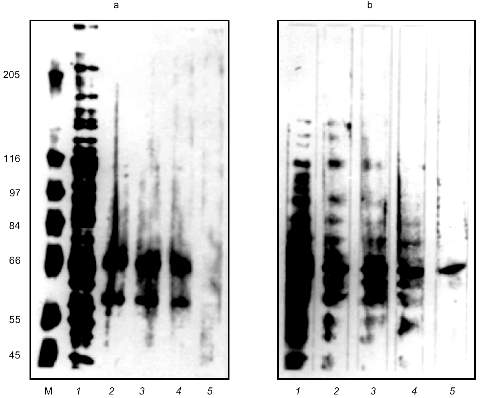
The eluted proteins were precipitated with acetone and analyzed by SDS-PAGE (Fig. 2a). The order of the sample loading is the same on Figs. 1b and 2a. There are two proteins with molecular masses of about 66 and 60 kD on lanes with eluted proteins loaded. Decreasing amounts of these proteins coincided with decreasing Alu-binding activity on corresponding lanes in the GMSA test (Fig. 1b). The presence of plasmid pAL as competitor during chromatography caused the absence of Alu-binding activity in the eluate (data not shown) and the absence of the 66- and 60-kD bands on SDS-PAGE (Fig. 2a, lane 5).
The southwestern blot was used to determine which protein is the DNA-binding one. The result of the proteins of DEAE fraction binding to Alu repeats in the presence of increasing amounts of nonspecific competitive DNA is shown on Fig. 2b. This method revealed a 66-kD protein which binds Alu repeats in the presence of 400-fold excess of E. coli DNA. The 60-kD protein was not detected by this method. This could be due to the small amount of the protein in the DEAE fraction, but more likely this protein is bound to Alu repeats by protein--protein interactions. The southwestern blot revealed the same 66-kD protein in the nuclear extract (data not shown).Fig. 2. SDS-PAGE of the affinity purified proteins (a) and southwestern blot with the labeled Alu fragment (b). a) M, molecular mass markers as indicated on the left (in kD); 1) 5 µl of DEAE fraction; 2-4) proteins bound to Alu DNA during affinity chromatography in the presence of 200-, 500-, and 1000-fold excess of competitive DNA, respectively. Lanes 2-4 correspond to lanes 2-4 on Fig. 1b; 5) affinity chromatography was done in the presence of 100-fold excess of plasmid pAL. Silver staining. b) Each lane was loaded with 15 µl of DEAE fraction. The incubation mixture for southwestern blotting contained labeled Alu fragment and 0- (1), 10- (2), 60- (3), 200- (4), and 400-fold (5) excess of E. coli DNA.
Chesnokov and Schmid [17] described a 60-kD Alu-binding protein found in human sperm. A small amount of a 50-kD protein is also reported in their article. These proteins were revealed using affinity purification on double-strand oligomers of oligonucleotide 5´-GATCTGTAATCCCA-3´, which is a part of the Alu repeat and is located between the A and B boxes of RNA polymerase III promoter. We suggested that our proteins and the proteins described in [17] are homologous. Antibodies to 60-kD Alu-binding protein were not reported in paper [17], so the possibility remained to compare the binding sites. Complementary oligonucleotides 5´-GATCTGTAATCCCA-3´ and 5´-GATCTGGGATTACA-3´ were synthesized. These oligonucleotides were annealed, labeled, and used in GMSA. It appear to form a specific complex with proteins of the DEAE fraction (Fig. 3a).
Alu were cleaved with Hae III in order to prove that the proteins bind the same sequence in Alu repeats of clone Blur8, which is the essential probe of the current work. The restriction sites in Alu repeats of clone Blur8 are shown in Fig. 4 (sequence identical to the synthetic oligonucleotide except two last nucleotides is underlined and bolded). Figure 4 show sequences of Alu repeats of clone Blur8 and consensus sequences of PV and major subfamilies. The sequence of clone Blur8 does not have the A box and its first 10 nucleotides are complementary to the oligonucleotide, so the Alu repeat of this clone was very suitable in this case. The left and right Hae III subfragments of Alu were applied to GMSA (Fig. 3, b and c). The proteins form complexes with both. The complexes are stable and do not disappear in the presence of a large amount of nonspecific competitive E. coli DNA. Addition of 50-fold molar excess of the nonlabeled oligonucleotide completely remove the complexes both in the case of labeled oligonucleotide and left Hae III Alu subfragment (lane 5 in Fig. 3, a and b). The retardation pattern of the left small Alu subfragment is identical to the one of the synthetic oligonucleotide because of size and nucleotide sequence similarity. The right big Alu subfragment also formed complexes according to previously published data [20]. This could be due to the right subfragment sequence homology to the synthetic oligonucleotide except in the third position (Fig. 4).Fig. 3. GMSA of DEAE active fraction proteins with synthetic oligonucleotide (a) and left (b) and right (c) Hae III Alu fragment subfragments. F, free labeled DNA. a) Mixtures with labeled oligonucleotide were incubated with 5 µl of the protein fraction in the presence of 0 (1), 60 (2), 200 (3), and 400 ng of plasmid pUC19 (4) and in the presence of 60 ng pUC19 and 50-fold excess of cold oligonucleotide (5); b) 1 ng of the labeled left Hae III Alu subfragment was incubated with 5 µl of DEAE fraction in the presence of 0 (1), 60 (2), 200 (3), and 400 ng of plasmid pUC19 (4) and in the presence of 60 ng pUC19 and 50-fold excess of cold oligonucleotide (5); c) 1 ng of the labeled right Hae III Alu subfragment was incubated with 5 µl of DEAE fraction in the presence of 0 (1), 60 (2), 200 (3), 400 (4), and 600 ng of plasmid pUC19 (5).
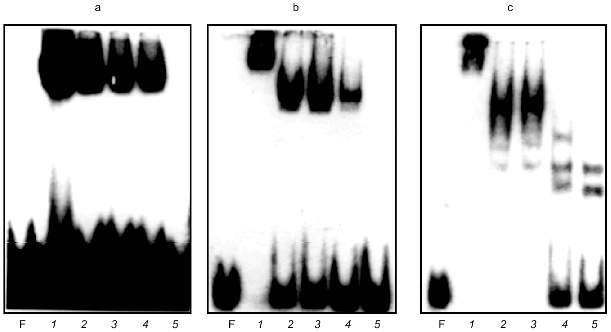
So, the 66-kD Alu-binding protein from somatic human cells is probably a homolog of the 60-kD protein from human sperm [17].Fig. 4. Alignment of the Alu-repeat sequence from the clone Blur8 (accession number J00091) and consensus sequences of the PV (HS) family (accession number U02043) and major family of Alu repeats [29]. The binding site of the 66-kD protein from the current paper and the Alu-binding protein from spermatozoids [17] is underlined and bolded. The binding site for the T-antigen and the proteins described in [16, 18, 20] are printed in italic. Arrows mark the sites of cleavage by restrictase Hae III. Open frames show the A and B boxes of the RNA polymerase III combined promotor. Shaded frames show the A-rich islets which separate the left and right monomers of the Alu repeat.

DISCUSSION
About two dozen articles devoted to Alu-binding proteins have been published to date. The reported results differ and sometimes contradict each other. The problem of determining the Alu-binding proteins characteristics is far of being solved. The Alu repeats are likely to be a collection of different binding sites beginning from the site for RNA polymerase III. So, a number of proteins could bind Alu repeats specifically despite the limited size of the sequence.
The 66-kD protein from nucleoplasm of placenta cells revealed in the present work specifically binds DNA of human Alu repeats. The affinity purification results show that the Alu repeats are also bound by the 60-kD protein. This binding is likely to be caused by protein--protein interactions. It was shown that the binding site of the 66-kD protein is situated between the A and B boxes and coincided with the site of Alu-binding protein from human sperm [17]. The authors of this article described a 60-kD Alu-binding protein which is specific for spermatozoid and is not revealed in the nuclear extract of HeLa cells. The two proteins of 66 and 60 kD revealed by affinity purification in the current work are likely to correspond to the two proteins with molecular masses of 60 and 50 kD which were revealed in spermatozoids by analogous affinity purification on oligonucleotides [17]. The 60-kD protein from somatic human tissues has no Alu-binding activity on southwestern blotting, unlike the 60-kD protein from spermatozoid [17]. According to the binding site revealed, it is most likely that the 66-kD protein from human somatic cells is homologous to the 60-kD protein from spermatozoids. Experimental data obtained and published are not enough to prove the identity of the two proteins, but similarity in isolation procedure and similarity of binding sites and character of DNA--protein complex formation suggest homology of the proteins. Interestingly, the protein revealed in somatic cells is not tissue specific and could be found in nuclear extract of human embryonal liver and HeLa cells. The question about homology or identity of Alu-binding proteins from somatic and germ cells could be solved with the help of antibodies.
A mechanism of transcriptional inhibition of Alu repeats has been proposed [22]; two proteins were found in nuclear extract of HeLa cells using GMSA on oligonucleotides. The proteins specifically bound the B box and a site just downstream from it. It was shown that the protein which binds the B box differs from TFIIIC and was not detected before. These proteins repress Alu repeat transcription as a result of binding. The molecular masses of these proteins were not defined. The proteins described in the current work were not tested especially in binding to these sites of the Alu repeats. However, it is to be noted that the proteins observed have high specificity to the oligonucleotide located between the A and B boxes, but sequences of the B box and the element located downstream from it differ in nucleotide composition (Fig. 4). Destruction of the correspond binding site by restrictase Hae III (Fig. 4, position 99) does not lead to disappearance of the DNA--protein complexes (Fig. 3, b and c).
Another potential binding site in Alu repeats is a conservative element of binding for large T antigen (GAGGCNGAGGC) located between the A and B boxes of Alu repeats [18]. Chesnokov et al. [15] found a protein which specifically binds the sequence AGGNNNAGG and AGG triplets with six nucleotides spacing between it using DNase I footprinting and GMSA with oligonucleotides. Southwestern blotting shows that it is a protein with molecular mass of 80 kD. Both the binding site and molecular mass of this protein differ from the protein described in the current paper.
Chiang and Vishwanatha found a 35-kD protein in nuclear extract of HeLa cells using labeled oligonucleotides which contained a large T antigen-binding site in southwestern blot analysis [16]. In the current work we destroyed the corresponding site GAGGCCAAGGAGGG in the Alu repeat of clone Blur8 (Fig. 4, positions 48-63) by restrictase Hae III (GGCC is the site of restriction). Nevertheless, the formation stable DNA--protein complexes with the left Alu subfragment in GMSA was observed (Fig. 3b).
The results of GMSA analysis suggest that the proteins revealed are probably heterodimer formed multimeric complexes with Alu repeats. The tendency to polymerization under conditions close to physiological may indicate that these proteins are components of some special cell structure. We hope to prove this with antibodies raised against these proteins.
The authors thank I. B. Lobov, N. I. Enukashvili, A. G. Urusov, and other members of our laboratory for the discussion of the results. This work was supported by grant from US DOE Human Genome Project and the program «Integration» from the Russian Government.
REFERENCES
1. Houck, C. M., Rinehard, F. P., and Schmid, C. W.
(1979) J. Mol. Biol., 132, 289-306.
2. Deininger, P. L. (1989) Mobile DNA.
3. Rogers, J. H. (1985) Nature, 317,
765-766.
4. Deininger, P. L., Batzer, M. A., Hutchison, C. A.,
and Edgell, M. H. (1992) Trends Genet., 8, 307-311.
5. Allan, M., and Paul, J. (1984) Nucleic Acids
Res., 12, 1193-1200.
6. Lim, D., Coleman, R. T., Assman, G., and Frossard,
P. M. (1986) Am. J. Hum. Genet., 39, 209.
7. Calos, M. P., and Miller, G. N. (1980)
Cell, 20, 579-595.
8. Jelinek, W. R., Toomey, T. P., Leinward, L.,
Duncan, C. H., Biro, P. A., Choudary, P., Weissman, S. M., Rubin, C.
M., Houck, C. M., Deininger, P. L., and Schmid, C. W. (1980) Proc.
Natl. Acad. Sci. USA, 77, 1398-1402.
9. Taylor, J. H. (1983) XV Int. Congr. of
Genetics, Oxford, pp. 1-2.
10. Korenberg, J. R., and Rykowski, M. C. (1988)
Cell, 53, 391-400.
11. Paulson, K. E., and Scmid, C. W. (1986)
Nucleic Acids Res., 14, 6145-6158.
12. Bovia, F., and Strub, K. J. (1996) Cell
Sci., 109, 2601-2608.
13. Chesnokov, I. N., and Scmid, C. W. (1996) J.
Mol. Evol., 42, 30-36.
14. Perelygina, L. M., Tomilin, N. V., and
Podgornaya, O. I. (1987) Mol. Biol. (Moscow), 21,
1610-1619.
15. Podgornaya, O. I., Perelygina, L. M., and
Tomilin, N. V. (1988) FEBS Lett., 232, 99-102.
16. Chiang, Y., and Vishwanatha, J. K. (1996)
Mol. Cell Biol., 155, 131-138.
17. Chesnokov, I. N., and Schmid, C. W. (1995) J.
Biol. Chem., 270, 18539-18542.
18. Tomilin, N. V., and Bozhkov, V. M. (1989)
FEBS Lett., 251, 79-83.
19. Tomilin, N. V., Iguchi-Ariga, S., and Ariga, H.
(1990) FEBS Lett., 263, 69-72.
20. Chesnokov, I. N., Bozhkov, V. M., Popov, B., and
Tomilin, N. V. (1991) Biochem. Biophys. Res. Commun.,
178, 613-619.
21. Tomilin, N. V., Bozhkov, V. M., Bradbury, E. M.,
and Schmid, C. W. (1992) Nucleic Acids Res., 20,
1941-2945.
22. Kropotov, A. V., and Tomilin, N. V. (1996)
Genetica, 98, 223-233.
23. Kropotov, A. V., and Tomilin, N. V. (1996)
FEBS Lett., 386, 43-46.
24. Berezney, R., and Coffey, D. S. (1974)
Biochem. Biophys. Res. Commun., 60, 1410-1417.
25. Rubin, C. M., Houck, C. M., Deininger, P. L.,
Friedmann, T., and Schmid, C. W. (1980) Nature, 284,
372-374.
26. Maniatis, T., Frich, E., and Sambrook, J. (1984)
Molecular Cloning [Russian translation], Mir, Moscow.
27. Strauss, F., and Varshavsky, A. (1984)
Cell, 37, 889-901.
28. Laemmli, U. K. (1970) Nature, 227,
680-682.
29. Towbin, H. T., Stahelin, T., and Cordon, J.
(1979) Proc. Natl. Acad. Sci USA, 76, 4350-4354.
30. Din, P., Johnson, W., and Middle, F. (1988)
Affinity Chromatography. Methods [Russian translation], Mir,
Moscow.