REVIEW: Oxidant--Nitric Oxide Signalling Mechanisms in Vascular Tissue
M. S. Wolin*, C. A. Davidson, P. M. Kaminski, R. P. Fayngersh, and K. M. Mohazzab-H.
Department of Physiology, New York Medical College, Valhalla, New York 10595, USA; fax: (914) 594-4826* To whom correspondence should be addressed.
Received April 6, 1998
Nitric oxide has several signalling mechanisms that can potentially control force generation by vascular smooth muscle. Some of these mechanisms include the stimulation of cGMP production by the soluble heme-containing form of guanylate cyclase (sGC), inhibition of mitochondrial respiration, and the modulation of vasoactive mediator release by the endothelium. Reactive O2 species (ROS) can also regulate force generation by vascular smooth muscle through mechanisms including the stimulation of production of vasoactive prostaglandins, the stimulation of sGC by catalase-mediated metabolism of H2O2 and inhibition of sGC activation by superoxide, the activation of protein kinase C, and the modulation of mediator release from the endothelium. Interactions between NO and ROS signalling mechanisms result in additional processes which modulate vascular force generation. For example, NO-elicited stimulation of sGC can be attenuated by superoxide, and this results in the formation of peroxynitrite (ONOO-). However, high levels of NO result in a ONOO- and thiol dependent formation of a species which regenerates NO in a time-dependent manner. It appears that NO inhibits catalase through an O2 and superoxide dependent process which results in inhibition of relaxation mediated by H2O2-elicited stimulation of sGC. Furthermore, evidence exists suggesting additional signalling mechanisms resulting from interactions between regulatory systems involving NO and ROS which appear to be important in control of vascular force generation in pathophysiological states.
KEY WORDS: endothelium-derived factors, guanylate cyclase, mitochondrial respiration, nitric oxide, oxidant signalling, peroxynitrite, prostaglandins, redox, thiol nitrosation, vascular signalling
Nitric oxide (NO) and oxidant signalling mechanisms have been implicated in many physiological and pathophysiological aspects of vascular function, including processes that control vascular force, the release of vasoactive mediators from endothelial cells, tissue respiration, inflammatory cell interactions, gene expression, and vessel wall remodeling. This review focuses on some of the better understood interactions between NO and oxidant second messenger-like signalling processes that appear to participate in the control of vascular force generation.
SIGNALLING MECHANISMS MEDIATED BY NITRIC OXIDE
Nitric oxide is known to have several specific signalling mechanisms of potential relevance to vascular function which are known to occur in the absence of an interaction with reactive O2 species (ROS). These mechanisms include stimulation of the soluble or cytosolic form of heme-containing guanylate cyclase (sGC) [1-3] and the reversible inhibition of cytochrome oxidase [4-6]. The stimulation of sGC by NO is known to occur through a binding of the ferrous heme present on this enzyme. When NO binds the heme of sGC, enzyme activation occurs as a result of the loss of binding of a histidine residue to the iron of the heme [1-3]. The mechanism through which NO inhibits cytochrome oxidase is thought to be mediated by a binding of a heme on this enzyme in a manner which is competitive with the binding of oxygen [4]. However, the actual process involved is not completely understood. The stimulation of sGC results in the production of cGMP. While cGMP is likely to have multiple actions of relevance to vascular function, its best documented action is to promote vascular relaxation. Studies on the mechanism through which cGMP causes vascular relaxation suggest a key role for activation of cGMP-dependent protein kinase, and the potential involvement of processes including inhibition of the release of calcium, stimulation of the reuptake of cellular calcium, and membrane hyperpolarization through the opening of potassium channels [7-9]. The inhibition of cytochrome oxidase in vascular tissue by NO would result in an impairment of energy metabolism, which is likely to result in vascular relaxation through membrane hyperpolarization as a result of the opening of ATP-dependent potassium channels and through a diminished availability of ATP that is required for contractile apparatus function. A large number of additional processes in vascular tissue have been linked to NO, including the cGMP-independent opening of potassium channels [10], the activation of p21ras [11], stimulation of prostaglandin-mediated signalling [12, 13], and the inhibition of proliferation [14]. While these processes are generally associated with thiol modification or nitrosation dependent mechanisms, it is not yet known if they are independent of interactions of NO with ROS.
SIGNALLING MECHANISMS MEDIATED BY OXIDANT SPECIES
Several vascular signalling mechanisms involving individual ROS species which are independent of an interaction with NO have been identified. Hydrogen peroxide (H2O2) is known to cause responses through stimulation of sGC [15-17], activation of protein kinase C [18, 19], prostaglandin production [20-23], and hyperpolarization [24] in various vascular preparations. In endothelium, H2O2 potentially modulates the release of vasoactive mediators through mechanisms including alterations in ion transport and calcium signalling [25-27], and by the stimulation of phospholipases C and D [28], whereas superoxide anion inhibits the stimulation of sGC by H2O2 and other stimuli [29]. The combination of superoxide and H2O2 results in the formation of hydroxyl radical-like species, and these species appear to promote the inhibition of uptake and the release of calcium from the sarcoplasmic reticulum in vascular smooth muscle [30]. While hydroxyl radical itself is probably too reactive to be a signalling molecule, peroxide-oxidized iron species stabilized by chelation are potentially regulatory agents which possess hydroxyl radical-like chemical reactivities.
One of the most sensitive mechanisms activated by H2O2 in vascular smooth muscle and endothelium is the production of prostaglandins. The prostaglandins produced vary with the vascular preparation examined and can result in either vasodilation or constriction. For example, H2O2 appears to cause an endothelium-dependent vasodilation of rat skeletal muscle arterioles (microvessels) in vivo, which is potentially mediated by prostaglandins E2 and I2 [22], whereas H2O2 causes an endothelium-independent contraction of human placental arteries and veins potentially mediated through thromboxane A2 [23]. In preparations in which H2O2 does not elicit prostaglandin-mediated responses or after responses of this type are eliminated with inhibitors of cyclooxygenase, a relaxation response that appears to be mediated by stimulation of sGC and cGMP is often observed [21, 31, 32]. The mechanism of stimulation of sGC by H2O2 appears to be a result of its metabolism by catalase, and catalase stimulates sGC through its compound I intermediate [15-17]. This mechanism of sGC regulation by H2O2 appears to function as a sensor of changes in O2 tension in certain vascular preparations as a result of the effect of O2 on cellular levels of H2O2 [16, 33-35]. In addition, vascular relaxation to H2O2 is attenuated by inhibition of Cu,Zn-SOD which results from a pretreatment with the copper chelator diethyldithiocarbamate [29]. While H2O2 has been reported to cause hyperpolarization of vascular smooth muscle [24], it is not known if cGMP or prostaglandins participate in this response. However, it is possible that H2O2 can cause hyperpolarization through the oxidation of thiols that are known to be present on potassium channels [36]. Several studies have identified a role for the stimulation of protein kinase C in contractile responses to H2O2 [18, 19]. However, the signalling mechanism that results in the activation of protein kinase C needs to be elucidated. The mechanism of release and inhibition of calcium uptake by sarcoplasmic reticulum by hydroxyl radical-like species is a poorly understood process which may involve the oxidation of thiols thought to be present on these proteins. Superoxide and other oxidants are potential inhibitors of mitochondrial functions [37]. The inhibition of mitochondrial function by these species is likely to depress force generation through mechanisms similar to those previously described for NO.
It has been observed that H2O2 has several previously mentioned effects which could potentially regulate mediator release from the endothelium. Some of the best known effects are the stimulation of prostaglandin production and promoting an increase cellular calcium which could result in the release of NO and perhaps other mediators. Thus, individual ROS have specific mechanisms of signalling which depend on their cellular levels in the endothelium and smooth muscle of the vessel wall.
CONTROL OF THE PRODUCTION AND INTERACTION OF NITRIC OXIDE AND
OXIDANT SPECIES IN VASCULAR TISSUE
Endothelium contains a calcium-regulated form of nitric oxide synthase (eNOS). This enzyme produces NO as a result of activation of the endothelium by receptor mediated stimuli, and the shear force of blood flow is often a stimulus for the release of NO [38]. If H2O2 causes an increase in endothelial cell calcium, this can also be a stimulus for NOS activation [25-27]. Vascular force generation can also be regulated from NO derived from a similar calcium-regulated form of NOS present in nerves. Inflammatory mediators can result in the expression of the inducible form of NOS in both the endothelium and vascular smooth muscle. This calcium-independent form of the enzyme appears to continuously produce NO in the absence of additional stimulation. In addition, inflammatory cells such as macrophages appear to be able to produce cytotoxic levels of NO-derived species.
Sources of ROS that can alter vascular function include inflammatory cells and systems present in vascular smooth muscle and endothelium. Inflammatory cells contain a receptor regulated NADPH oxidase which is able to produce cytotoxic levels of ROS [39]. While endothelium and smooth muscle have been observed to contain detectable levels of NADPH oxidase activity, it is not known if this is the same oxidase as the one present in inflammatory cells. Unstimulated endothelium and vascular smooth muscle have been observed to contain a microsomal NADH oxidase whose activity seems to be linked to the availability of O2 and cytosolic NADH, making this oxidase a potential pO2 and metabolic sensor [34, 35, 40]. This NADH oxidase has a cytochrome b558 and a p21 subunit which are very similar to the phagocytic oxidase [40, 41]. The activation of certain receptors, such as the angiotensin type I receptor, have been observed to increase the activity and expression of this oxidase [41, 42]. While the origin of vascular NADPH oxidase activity is less well studied, it can potentially originate from the use of NADPH as a substrate for the NADH oxidase and from enzymes including cytochrome P-450 and NOS. In fact, endothelial NOS has been observed to be a source of vascular relaxant levels of H2O2 under conditions of lowered levels of its cofactor tetrahydrobiopterin [43]. Endothelial xanthine oxidase and cyclooxygenase are two other systems which have also been demonstrated to produce vasoactive levels of ROS [44, 45]. Cyclooxygenase produces superoxide through the co-oxidation of cofactors such as NAD(P)H [46]. Endothelial xanthine dehydrogenase seems to require conversion to its oxidase form by processes such as endothelial perturbation produced by exposure to hypoxia followed by reoxygenation [47] before it can produce vasoactive levels of ROS. While mitochondria are considered to be a major source of production of ROS in certain cell types, there is little evidence to suggest that they are an important source of ROS in vascular tissue [35, 48].
The direct interaction of NO with ROS such as superoxide appears to be an important aspect of vascular signalling processes. This results primarily from the extreme efficiency of the chemical reaction between superoxide and NO, which has a bimolecular rate constant of 7·109 M-1·sec-1 [49, 50]. Since the rate of reaction of NO with superoxide is 3-fold greater than rate of reaction with SOD, NO will compete with SOD for the trapping of superoxide as its concentrations approach the nanomolar levels of SOD normally present in cells. The product of this reaction, peroxynitrite (ONOO-), appears to be detectable in endothelium intact blood vessels even in the absence of stimulation [51, 52]. Based on the diffusion properties of NO and the dominant sites of production of superoxide being in the intracellular environment of the vessel wall, it is likely that both endothelium and vascular smooth muscle are normally exposed to low levels of ONOO-, which potentially results from endothelium-derived NO interacting with superoxide produced in both of these cell types. As NO levels increase over the nanomolar concentration range it will scavenge superoxide and divert it from producing H2O2. This will produce an attenuation of ROS-dependent signalling and activation of mechanisms that result from the production of ONOO-. The more extensively studied interaction between NO and superoxide is the attenuation of NO-dependent processes, such as vasodilation, which occurs as a result of elevated levels of superoxide scavenging appreciable amounts of the NO that is present. There is now substantial evidence that many vascular diseases are associated with increased levels of production of superoxide, and perhaps also with decreased levels of SOD [53-56]. In some vascular diseases the production of both NO and superoxide seem to be quite elevated, and this condition should favor signalling and injury processes that result from ONOO-.
There also appear to be regulatory processes that result from effects of NO and ROS on each others signalling mechanisms, in addition to the chemical interactions between NO and superoxide. For example, the inhibition of mitochondrial function by NO-dependent mechanisms could result in increased production of ROS [5, 57, 58], and NO also appears to inhibit catalase and its role in the H2O2-dependent stimulation of sGC [59]. Since the interactions between NO and ROS signalling systems are likely to be depend the concentrations of each species present relative to its potential for signalling effects, careful consideration needs to be given to the levels of each species present.
MODULATION OF NO SIGNALLING BY ROS
The initial observations that superoxide inactivated endothelium-dependent relaxation had a major influence on establishing that the endothelium was able to produce NO [60, 61]. Cu,Zn-SOD activity in the endothelium and vascular smooth muscle appears to be essential for the release of NO and its relaxant actions, respectively [29, 62, 63]. An extracellular form of Cu,Zn-SOD present in the vessel wall also appears to be an important contributor to the protection of NO from inactivation by endogenous superoxide production [64]. Since NO-elicited cGMP-mediated relaxation seems to occur over the low nanomolar range, similar low nanomolar levels of superoxide will attenuate NO-dependent responses. Thus, the attenuation of NO-elicited stimulation of sGC seems to be a very important signalling mechanism for superoxide, which is likely to participate in many pathophysiological vascular responses. In some instances [65] NO production and/or NOS activity appear to be increased in vascular pathophysiological models associated with increased ROS, and this can normalize endothelium-dependent relaxation responses mediated by NO.
The reversible inhibition of mitochondrial respiration in tissues by NO is converted to an irreversible inhibition as a result of the interaction of NO with superoxide [66, 67]. It seems that under these conditions the intracellular formation of ONOO- may be the key process through which mitochondrial function is altered [67]. In bovine cardiac muscle, exposure to hypoxia + reoxygenation causes a ONOO- mediated inhibition of respiration involving cyclooxygenase and xanthine oxidase derived sources of superoxide which are likely to originate from the vascular endothelium [67, 68]. Thus, it appears that the vascular endothelium can also be a key source of levels of superoxide which participate in the inhibition of tissue respiration through the intracellular formation of ONOO-.
Hydrogen peroxide has been observed to cause an endothelium-dependent and prostaglandin-independent relaxation of rabbit aorta [26]. This response is likely to originate from an elevation of endothelial cell calcium by H2O2, which could participate in the stimulation of NOS [27]. In the cerebral microcirculation, H2O2 has also been reported to inhibit responses that seem to be mediated through endogenous NO-related signalling [69]. Thus, it appears that ROS can also modulate endothelial cell release of NO-related species through their effects on endothelial cell signalling.
MODULATION OF OXIDANT SIGNALLING BY NITRIC OXIDE
Nitric oxide has multiple interactions with ROS-related signalling mechanisms. First, it is such a potent scavenger of superoxide that it can compete with SOD for the metabolism of NO as the levels of NO approach the tissue levels of SOD. Based on our studies in the bovine pulmonary and coronary artery, 50 nM NO scavenges approximately 50% of the intracellular superoxide that is detectable with lucigenin chemiluminescence [70, 71]. Thus, as NO levels increase through the nanomolar range it will function as an intracellular scavenger of superoxide and this will shift the signalling mechanisms activated by ROS over to alternative mechanisms activated by NO-derived species. An additional potent site of action of NO is the inhibition of catalase [59, 72]. Our studies suggest that exposure of bovine pulmonary arteries to 50 nM NO for 2 min results in an inhibition of catalase and H2O2- and cGMP-mediated relaxing mechanisms that are dependent of the metabolism of peroxide by catalase [59]. Additional sites which NO is likely to alter signalling through ROS is by interactions with systems that produce mediators (such as prostaglandins) from the metabolism of arachidonic acid. For example, prostaglandin production is dependent on the availability of arachidonic acid and peroxide, where the availability of both of these species could be modulated by NO. In addition, NO-derived species appear to have interactions with many of the arachidonic acid metabolizing enzymes [73-75]. For example, ONOO- is an inhibitor of prostacyclin synthase [75] and this action could have dramatic effects on vascular responses mediated through prostaglandins, such as converting a vasodilator response to vasoconstriction.
NOVEL SIGNALLING MECHANISMS ORIGINATING FROM OXIDANT--NITRIC
OXIDE INTERACTIONS
In this section signalling mechanisms derived from oxidant--NO interactions will be further discussed. The dominant effect of ONOO- on bovine pulmonary arterial smooth muscle is to cause the formation of tissue species which release NO over a prolonged period of time [76, 77]. Since this was shown to be a thiol-dependent process, it is likely that nitrosated or nitrated thiols are important participants in the formation of NO. The data in Fig. 1 shows that a 15 min pretreatment of bovine pulmonary arterial smooth muscle with a the dose of a NO donor that produces maximal vascular relaxation only causes an inhibition of force generation to serotonin (after removal of the NO donor) in the presence of a source of superoxide generation. A 2 min exposure to approximately 50 nM levels of NO was employed to examine if the effects of endogenously generated ONOO- resembled the actions of exogenous ONOO-, since, as previously mentioned, exposure of bovine vascular tissue to this level of NO caused a substantial scavenging of intracellular superoxide. The data in Fig. 2 show that as the concentration of NO used during the 2 min exposure increases from 25 to 50 nM a prolonged relaxation resembling the response previously seen with exogenous ONOO- [76] was also observed. The prolonged relaxation evoked by 50 nM NO was previously observed [70] to be inhibited by the presence of an intracellular scavenger of superoxide (10 mM Tiron) during the NO exposure and by post NO treatment with a scavenger of NO (1 µM hemoglobin) and with an inhibitor of the sGC stimulating action of NO (10 µM methylene blue). In addition, the NO treatment was also observed to cause a thiol dependent trapping of NO [70]. The data in Fig. 3 show that if 0.1 mM urate, an intracellular scavenger of ONOO- [67], is present during the exposure to 50 nM NO, it causes an attenuation of the prolonged relaxation. Thus, exposure of bovine pulmonary arterial smooth muscle to 50 nM NO produces the formation of an amount of ONOO- which approximately mimics the effects of exposure to exogenous 0.1 mM ONOO-. These data suggest that ONOO- formation is likely to occur in tissues at elevated biological levels of NO, and an important effect of this level of ONOO- is the trapping of NO by a thiol dependent process which is likely to involve the formation of endogenous NO-thiol species.
Fig. 1. Effects of a 15 min pretreatment of endothelium-removed bovine pulmonary arteries with 10 µM of the NO donor S-nitroso-N-acetylpenicillamine (NO) (3), the superoxide-generating system xanthine oxidase (10 mU/ml) + hypoxanthine (1 mM) + catalase (10 µM) (O2.-) (2), and the ONOO--generating system NO + O2.- (4) on the cumulative concentration-dependent contraction to increasing doses of serotonin after washout of the NO- and O2.--generating systems (curve (1) is control). The vascular preparation used in the experiments shown in Figs. 1-5 is described in paper [59] (n = 7, ANOVA Statistics, mean ± SEM); *p < 0.05.
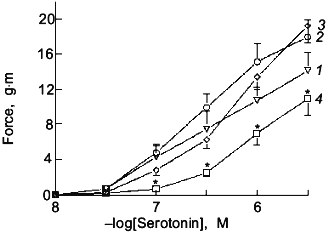
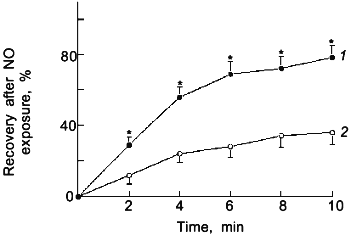
Fig. 2. Time course for the recovery of force generation to 30 mM KCl after a 2 min exposure of endothelium-removed bovine pulmonary arteries to 25 (1) and 50 nM NO (2) (n = 6); *p < 0.05.
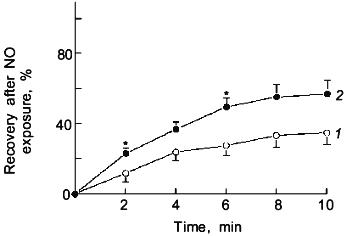
The previously mentioned inhibition of catalase in bovine pulmonary arterial smooth muscle by the 2 min treatment with 50 nM NO [59] could involve an effect of NO or its oxidized species, such as ONOO-. The data in Fig. 4 indicates that the inhibition of relaxation to H2O2 caused by the 2 min exposure to 50 nM NO is not observed when the NO exposure and the subsequent relaxation response to H2O2 are examined under a severe hypoxia (pO2 = 8-10 torr) produced by a N2 (5% CO2) containing atmosphere. As shown in Fig. 5, the presence of an intracellular scavenger of superoxide during exposure to 50 nM NO markedly attenuated the effect of this NO treatment on the previously observed inhibition of bovine pulmonary artery relaxation to endogenously formed H2O2.Fig. 3. Effects of the presence of 0.1 mM urate (ONOO- scavenger) on the time course for the recovery of force generation to 30 mM KCl after a 2 min exposure of endothelium-removed bovine pulmonary arteries to 50 nM NO (n = 7): 1) control; 2) anions of uric acid; *p < 0.05.
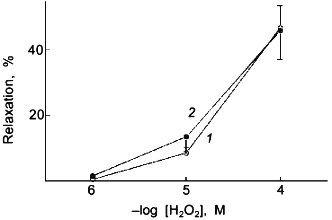
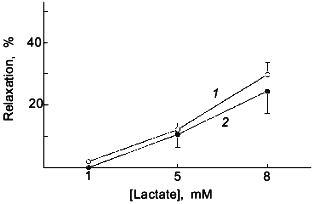
Lactate was employed to promote a relaxation through increased H2O2 in bovine pulmonary artery smooth muscle as a result of its effect on elevating the levels of cytosolic NADH by the lactate dehydrogenase reaction [40, 78]. It was previously observed that the treatment of purified catalase with 50 nM NO for 2 min inhibited its activity [59]. However, Tiron did not prevent this effect of NO on catalase and the exposure of purified catalase to 0.1 mM ONOO- or 67 µM NO2 did not inhibit its activity (data not shown). While a role for ONOO- is suggested in the inhibition of arterial catalase by the 2 min treatment with NO, the actual species of NO that inhibits catalase is not known. It is possible that the tonic release of NO that results from ONOO- formation during the NO treatment participates in the inhibition of catalase and vascular responses dependent on H2O2 metabolism by catalase.
Fig. 4. The previously observed [59] attenuation by a 2 min pretreatment with 50 nM NO (2) ((1) is control (N2)) of relaxation of KCl-precontracted endothelium-removed bovine pulmonary arteries to increasing cumulative concentrations of H2O2 is not observed when this experiment is conducted under a severely hypoxic atmosphere produced by gassing with 95% N2 + 5% CO2 (n = 5).
It is likely that additional vascular signalling mechanisms will result from the interaction of NO with superoxide because the product of this reaction, ONOO-, can potentially regulate other systems through its ability to modify thiols on key components of these systems or as a result of its prolonged inhibitory action on mitochondrial respiration. The enzymes involved in the production of prostaglandins [73-75], the inhibition of tyrosine phosphatases [79], the function of potassium channels [10], and signalling dependent on the actions of p21ras [11] are examples of systems already identified which can be regulated by NO-elicited thiol modification processes. Thus, we speculate that the modification of key thiol groups by ONOO--dependent processes is potentially a fundamental signalling process involved in important mechanisms of control of vascular force generation.Fig. 5. The previously observed [59] attenuation by a 2 min pretreatment with 50 nM NO (2) of relaxation of KCl precontracted endothelium-removed bovine pulmonary arteries to endogenously generated H2O2 elicited by increasing cumulative concentrations of lactate is not observed when this experiment is conducted in the presence of an intracellular scavenger of superoxide, 10 mM Tiron ((1) is control) (n = 5).
RELEVANCE OF OXIDANT--NITRIC OXIDE SIGNALLING MECHANISMS IN
VASCULAR PATHOPHYSIOLOGY
Evidence already exists for the importance of alterations in NO-superoxide signalling in vascular pathophysiology associated with atherosclerosis [54, 56], hypertension [53, 56], diabetes [55], tissue ischemia-reperfusion [80], and many other disease processes. Thus, the signalling mechanisms discussed here and additional processes which remain to be identified that are speculated to originate from the modification of key thiol groups by oxidant--NO interactions are likely to be regulatory processes that are of great importance in vascular pathophysiology.
This work was supported by USPHS Grants HL31069 and HL43023.
REFERENCES
1.Wolin, M. S., Wood, K. S., and Ignarro, L. J.
(1982) J. Biol. Chem., 257, 13312-13320.
2.Stone, J. R., and Marletta, M. A. (1994)
Biochemistry, 33, 5636-5640.
3.Yu, A. E., Hu, S., Spiro, T. G., and Burstyn, J. N.
(1994) J. Am. Chem. Soc., 116, 4117-4118.
4.Brown, G. C., and Cooper, C. E. (1994) FEBS
Lett., 356, 295-298.
5.Cleeter, M. W. J., Cooper, J. M., Darley-Usmar, V.
M., Moncada, S., and Schapira, A. H. V. (1994) FEBS Lett.,
345, 50-54.
6.Schweizer, M., and Richter, C. (1994) Biochem.
Biophys. Res. Commun., 204, 169-175.
7.Lincoln, T. M., Komalavilas, P., and Cornwell, T.
L. (1994) Hypertension, 23, 1141-1147.
8.Liu, H., Xiong, Z., and Sperelakis, N. (1997) J.
Mol. Cell. Cardiol., 29, 1411-1421.
9.Archer, S. L., Huang, J. M. C., Hampl, V., Nelson,
D. P., Shultz, P. J., and Weir, E. K. (1994) Proc. Natl. Acad. Sci.
USA, 91, 7583-7587.
10.Bolotina, V. M., Najibi, S., Palacino, J. J.,
Pagano, P. J., and Cohen, R. A. (1994) Nature, 368,
850-853.
11.Lander, H. M., Hajjar, D. P., Hempstead, B. L.,
Mirza, U. A., Chait, B. T., Cambell, S., and Quilliam, L. A. (1997)
J. Biol. Chem., 272, 4323-4326.
12.Salvemini, D., Misko, T. P., Masferrer, J. L.,
Siebert, K., Currie, M. G., and Needleman, P. (1993) Proc. Natl.
Acad. Sci. USA, 90, 7240-7244.
13.Inoue, T., Fukuo, K., Morimoto, S., Koh, E., and
Ogihara, T. (1993) Biochem. Biophys. Res. Commun., 194,
420-424.
14.Cornwell, T. L., Arnold, E., Boerth, N. J., and
Lincoln, T. M. (1994) Am. J. Physiol., 267,
C1405-C1413.
15.Burke, T. M., and Wolin, M. S. (1987) Am. J.
Physiol., 252, H721-H732.
16.Burke-Wolin, T. M., and Wolin, M. S. (1990)
Am. J. Physiol., 258, H1267-H1273.
17.Cherry, P. D., and Wolin, M. S. (1989) Free
Rad. Med. Biol., 7, 485-490.
18.Packer, C. S., and Rhoades, R. A. (1992) Lung
Biol. Health Dis., 60, 227-262.
19.Rao, G. N., Runge, M. S., and Alexander, R. W.
(1995) Biochim. Biophys. Acta, 1265, 67-72.
20.Gurtner, G. H., and Burke-Wolin, T. (1991) Am.
J. Physiol., 260, L207-L211.
21.Burke-Wolin, T., Abate, C. J., Wolin, M. S., and
Gurtner, G. H. (1991) Am. J. Physiol., 261,
L393-L398.
22.Wolin, M. S., Rodenberg, J. M., Messina, E. J.,
and Kaley, G. (1987) Am. J. Physiol., 252,
H1159-H1163.
23.Omar, H. A., Figueroa, R., Omar, R. A., Tejani,
N., and Wolin, M. S. (1992) Am. J. Obstet. Gynecol., 167,
201-207.
24.Weir, E. K., and Archer, S. L. (1995) FASEB
J., 9, 183-189.
25.Elliott, S., and Koliwad, S. K. (1995) Free
Rad. Biol. Med., 19, 649-658.
26.Zembowicz, A., Hatchett, R. J., Jakubowski, A.
M., and Gryglewski, R. J. (1993) Br. J. Pharmacol., 110,
151-158.
27.Shimizu, S., Ishii, M., Yamamoto, T., and Momose,
K. (1997) Res. Commun. Mol. Pathol. Pharmacol., 95,
227-239.
28.Natarajan, V. (1995) J. Lab. Clin. Med.,
125, 26-37.
29.Cherry, P. D., Omar, H. A., Farrell, K. A.,
Stuart, J. S., and Wolin, M. S. (1990) Am. J. Physiol.,
259, H1056-H1062.
30.Grover, A. K., Samson, S. E., Fomin, V. P., and
Werstiuk, E. S. (1995) Am. J. Physiol., 269,
C546-C553.
31.Wolin, M. S., Rodenburg, J. M., Messina, E. J.,
and Kaley, G. (1990) J. Pharmacol. Exp. Ther., 253,
508-512.
32.Omar, H. A., Figueroa, R., Tejani, N., and Wolin,
M. S. (1993) Am. J. Obstet. Gynecol., 169, 912-918.
33.Burke-Wolin, T. M., and Wolin, M. S. (1989) J.
Appl. Physiol., 66, 167-170.
34.Mohazzab-H., K. M., Fayngersh, R. P., and Wolin,
M. S. (1995) Am. J. Physiol., 269, L637-L644.
35.Mohazzab-H., K. M., Kaminski, P. M., Fayngersh,
R. P., and Wolin, M. S. (1996) Am. J. Physiol., 270,
H1044-H1053.
36.Duprat, F., Guillemare, E., Romey, G., Fink, M.,
Lesage, F., and Lazdunski, M. (1995) Proc. Natl. Acad. Sci. USA,
92, 11796-11800.
37.Gardner, P. R., Nguyen, D. H., and White, C. W.
(1994) Proc. Natl. Acad. Sci. USA, 91, 12248-12252.
38.Sase, K., and Michel, T. (1997) Trends
Cardiovasc. Med., 7, 28-37.
39.DeLeo, F. R., and Quinn, M. T. (1996) J.
Leukoc. Biol., 60, 677-691.
40.Mohazzab-H., K. M., and Wolin, M. S. (1994)
Am. J. Physiol., 267, L823-L831.
41.Ushio-Fukai, M., Zafari, A. M., Fukui, T.,
Ishizaka, N., and Griendling, K. K. (1996) J. Biol. Chem.,
271, 23317-23321.
42.Griendling, K. K., Minieri, C. A., Ollerenshaw,
J. D., and Alexander, R. W. (1994) Circ. Res., 74,
1141-1148.
43.Cosentino, F., and Katusic, Z. S. (1995)
Circulation, 91, 139-144.
44.Ohara, Y., Peterson, T. E., and Harrison, D. G.
(1993) J. Clin. Invest., 91, 2546-2551.
45.Marshall, J. J., and Kontos, H. A. (1991) in
Cardiovascular Significance of Endothelium-Derived Vasoactive
Factors (Rubanyi, G., ed.) Futura Publishing Co, Mount Kisco, New
York, pp. 125-145.
46.Kukreja, R. C., Kontos, H. A., Hess, M. L., and
Ellis, E. F. (1986) Circ. Res., 59, 612-619.
47.Granger, D. N. (1988) Am. J. Physiol.,
255, H1269-H1275.
48.Mohazzab-H., K. M., and Wolin, M. S. (1994)
Am. J. Physiol., 267, L815-L822.
49.Beckman, J. S. (1996) Chem. Res. Toxicol.,
9, 836-844.
50.Pryor, W. A., and Squadrito, G. L. (1995) Am.
J. Physiol., 268, L699-L722.
51.Kooy, N. W., and Royall, J. A. (1994) Arch.
Biochem. Biophys., 310, 352-359.
52.Wolin, M. S., and Mohazzab-H., K. M. (1997) in
Oxidative Stress and the Molecular Biology of Antioxidant
Defenses (Scandalios, J. G., ed.) Cold Spring Harbor Laboratory
Press, Cold Spring Harbor, New York, pp. 21-48.
53.Wei, E. P., Kontos, H. A., Christman, C. W.,
DeWitt, D. S., and Povlishock, J. T. (1985) Circ. Res.,
57, 781-787.
54.Mugge, A., Elwell, J. H., Peterson, T. E.,
Hofmeyer, T. G., Heistad, D. D., and Harrison, D. G. (1991) Circ.
Res., 69, 1293-1300.
55.Tesfamariam, B. (1994) Free Rad. Biol.
Med., 16, 383-391.
56.Alexander, R. W. (1995) Hypertension,
25, 155-161.
57.Poderoso, J. J., Carreras, M. C., Lisdero, C.,
Riobo, N., Schopfer, F., and Boveris, A. (1996) Arch. Biochem.
Biophys., 328, 85-92.
58.Mohazzab-H., K. M., Xie, Y.-W., and Wolin, M. S.
(1997) FASEB J., 11, A644.
59.Mohazzab-H., K. M., Fayngersh, R. P., and Wolin,
M. S. (1996) Am. J. Physiol., 271, H1900-H1906.
60.Furchgott, R. F., Khan, M. T., and Jothianandan,
K. D. (1990) in Endothelium-Derived Relaxing Factors (Rubanyi,
G. M., and Vanhoutte, P. M., eds.) S. Karger, Basal, pp. 8-21.
61.Ignarro, L. J. (1989) Circ. Res.,
65, 1-21.
62.Mugge, A., Elwell, J. H., Peterson, T. E., and
Harrison, D. G. (1991) Am. J. Physiol., 260,
C219-C225.
63.Omar, H. A., Cherry, P. D., Mortelliti, M. P.,
Burke-Wolin, T., and Wolin, M. S. (1991) Circ. Res., 69,
601-608.
64.Abrahamsson, T., Brandt, U., Marklund, S. L., and
Sjoqvist, P.-O. (1992) Circ. Res., 70, 264-271.
65.Cosentino, F., Hishikawa, K., Katusic, Z. S., and
Luscher, T. F. (1997) Circulation, 96, 25-28.
66.Radi, R., Rodriguez, M., Castro, L., and Telleri,
R. (1994) Arch. Biochem. Biophys., 308, 89-95.
67.Xie, Y.-W., and Wolin, M. S. (1996)
Circulation, 94, 2580-2586.
68.Wolin, M. S., Hintze, T. H., Shen, W.,
Mohazzab-H., K. M., and Xie, Y.-W. (1997) Biochem. Soc. Trans.,
in press.
69.Wei, E., and Kontos, H. A. (1990)
Hypertension, 16, 162-169.
70.Davidson, C. A., Kaminski, P. M., and Wolin, M.
S. (1997) Am. J. Physiol., in press.
71.Davidson, C. A., Kaminski, P. M., and Wolin, M.
S. (1997) Nitric. Oxide Biol. Med., in press.
72.Brown, G. C. (1995) Eur. J. Biochem.,
232, 188-191.
73.Landino, L. M., Crews, B. C., Timmons, M. D.,
Morrow, J. D., and Marnett, L. J. (1996) Proc. Natl. Acad. Sci.
USA, 93, 15069-15074.
74.Gunther, M. R., Hsi, L. C., Curtis, J. F.,
Gierse, J. K., Marnett, L. J., Eling, T. E., and Mason, R. P. (1997)
J. Biol. Chem., 272, 17086-17090.
75.Zou, M.-H., and Ullrich, V. (1996) FEBS
Lett., 382, 101-104.
76.Wu, M., Pritchard, K. A., Kaminski, P. M.,
Fayngersh, R. P., Hintze, T. H., and Wolin, M. S. (1994) Am. J.
Physiol., 266, H2108-H2113.
77.Davidson, C. A., Kaminski, P. M., Wu, M., and
Wolin, M. S. (1996) Am. J. Physiol., 270,
H1038-H1043.
78.Omar, H. A., Mohazzab-H., K. M., Mortelliti, M.
P., and Wolin, M. S. (1993) Am. J. Physiol., 264,
L141-L145.
79.Caselli, A., Chiarugi, P., Camici, G., Manao, G.,
and Ramponi, G. (1995) FEBS Lett., 374, 249-252.
80.Nonami, Y. (1997) Jpn. Circ. J.,
61, 119-132.