REVIEW: Phosphoinositide Metabolism and Ca2+ Oscillation
V. A. Tkachuk
Department of Biological and Medical Chemistry, School of Basic Medicine, Lomonosov Moscow State University, Moscow, 119899 Russia
Received August 6, 1997
The main pathways of hydrolysis and synthesis of phosphoinositides, metabolism of inositol phosphates and their role in animal and human cells are reviewed. Mechanisms of regulation of phospholipase C and other key enzymes of phosphoinositide metabolism by hormones and growth factors are discussed. The mechanisms of regulation of metabolism and cellular activity by second messengers formed during activation of phosphoinositide hydrolysis are considered. Special attention is given to the regulation of cytoplasmic Ca2+ level by inositol phosphates. The review summarizes data on formation and possible biological role of inositol phosphates and phosphoinositides containing phosphate at position 3. The role of phosphoinositides in the interaction of cellular membranes with proteins of the cytoskeleton is considered.
KEY WORDS: phosphoinositide metabolism, calcium, hormones, growth factors
Abbreviations: Ins-4-P) inositol-4-monophosphate; Ins-4,5-P2) inositol-4,5-bisphosphate; Ins-1,4,5-P3) inositol-1,4,5-trisphosphate; Ins-1,3,4,5-P4) inositol-1,3,4,5-tetrakisphosphate; PI) monophosphoinositide; PIP) diphosphoinositide; PIP2) triphosphoinositide; DAG) diacylglycerol; PI-4,5-P2) phosphatidylinositol-4,5-bisphosphate; PI-4-P) phosphatidylinositol-4-monophosphate; PA) phosphatidic acid; PDGF) platelet-derived growth factor.
The story of the role of phosphoinositide metabolism in hormonal signal
transduction began from the discovery by Hokin's couple. They found
that acetylcholine increased incorporation of radiolabeled phosphate
into pancreatic phospholipids [1], mainly into
phosphatidylinositol and phosphatidic acid (PA) [2]. Robert Michell noticed that hormones increasing
Ca2+ influx into cells activate phosphoinositide metabolism,
and he suggested that activation of phosphoinositide hydrolysis leads
to the opening of plasma membrane calcium channels [3]. In fact, Ca2+ mobilizing hormones
increase intracellular levels of inositol-4,5-bisphosphate
(Ins-4,5-P2) and inositol-1,4,5-trisphosphate
(Ins-1,4,5-P3) in a few seconds [4].
Kinetics of their formation correlated with Ca2+ release
from intracellular stores and influx of extracellular Ca2+
into the cytoplasm, and Berridge suggested that Ins-1,4,5-P3
mobilizes Ca2+ in cells [4]. In the same
year (1983) Ca2+ release from its intracellular stores by
Ins-1,4,5-P3 was demonstrated [5]. In a
short time intracellular formation of another inositol-trisphosphate
(Ins-1,3,4-P3) [6, 7] and its precursor,
inositol-1,3,4,5-tetrakisphosphate (Ins-1,3,4,5-P4) [8], formed by phosphorylation of
Ins-1,4,5-P3 was demonstrated. Additional isomers of
InsP4 increased by hormones and high levels of
InsP5 and InsP6 (fitin) were also found in
various cells [9-14]. However,
hormones did not influence concentrations of two latter inositol
phosphates.
The total amount of inositol-containing phospholipids (phosphoinositides) varies in cellular membranes of different tissues from 2 to 8% [15] and phosphatidylinositol (PI) represents the highest proportion of them. Most phosphatidylinositol is located in the inner membranes, whereas phosphoinositides which contain more phosphate groups (phosphatidylinositol-4-phosphate, PI-4-P; phosphatidylinositol-4,5-bisphosphate, PI-4,5-P2) are preferentially located in the plasma membrane [16]. Phosphoinositides which are substrates for phospholipase C usually contain stearic (at the first position of glycerol) and arachidonic (at the second position of glycerol) acids.
Hydrolysis of triphosphoinositides which occurs on activation of phospholipase C yields two molecules. Hydrophilic Ins-1,3,4-P3 diffuses into the cytoplasm, whereas hydrophobic DAG remains in the membrane (Fig. 1). Ins-1,3,4-P3 binds to its receptors on the membrane of reticulum and in some cells on the cytoplasmic membrane. This results in opening of ions channels and increase of cytoplasmic Ca2+. Subsequent metabolism of Ins-1,3,4-P3 leads to complete dephosphorylation and formation of inositol. DAG activates protein kinase C and then is phosphorylated, i.e., is converted into phosphatidic acid. In the presence of CTP the latter is activated and condensed with inositol, forming monophosphoinositide. This phospholipid can be phosphorylated by various kinases resulting in formation of various isomers of diphospho- and triphosphoinositides (Fig. 1). Besides well known phosphoinositides containing phosphate at positions 1, 4, and 5, inositol-containing phospholipids that possess phosphate at position 3 have also been found. These are phosphatidylinositol-3-monophosphate, phosphatidylinositol-3,4-bisphosphate, and phosphatidylinositol-3,4,5-trisphosphate. Phosphoinositide kinase type I catalyzes their synthesis and differs from phosphoinositide kinase type II catalyzing phosphate incorporation into position 4 [17]. Kinase type I is believed to play an important role in the regulation of cell proliferation. Data on the activation of phosphoinositide kinase type I by platelet-derived growth factor (PDGF) accompanied by accumulation of polyphosphoinositides containing phosphate at position 3 of inositol support this hypothesis [18]. Mutant receptors of PDGF lacking the ability to bind kinase type I do not stimulate cell proliferation. Kinase type I is involved not only in cell proliferation. The formation of this reaction product was also detected in platelets during the action of adhesion inducers [19]. Triphosphoinositide possessing phosphate at positions 3, 4, and 5 is done by very specific and effective activators of one of the protein kinase C isoforms [20].
Protein kinase C discovered to be the main target for diacylglycerol formed during activation of phosphoinositide metabolism [21] is one of the best studied enzymes of this signal system. The family of protein kinase C isoforms is subdivided into three groups: (1) enzymes regulated by Ca2+ and diacylglycerol; (2) enzymes regulated by diacylglycerol only; (3) enzymes insensitive to either of these second messengers [22]. Products of hydrolysis of PIP2, phosphorylated at positions 1, 4, and 5 stimulate the first group of isomers of protein kinase C. Hydrolysis of phosphatidylcholine by phospholipases C and D causes activation of the second group of isoforms. PIP2 containing phosphate at positions 3, 4, and 5 and products of the ceramide pathway regulate the third group of isoforms of protein kinase C [23].Fig. 1. The main pathway of phosphoinositide metabolism. PI, PIP, and PIP2 are mono-, di-, and triphosphoinositide, respectively; Ins-4-P, Ins-1,4-P2, and Ins-1,4,5-P3 are inositol-mono-, inositol-bis-, and inositol-trisphosphate, respectively; DAG, diacylglycerol; PA, phosphatidic acid.
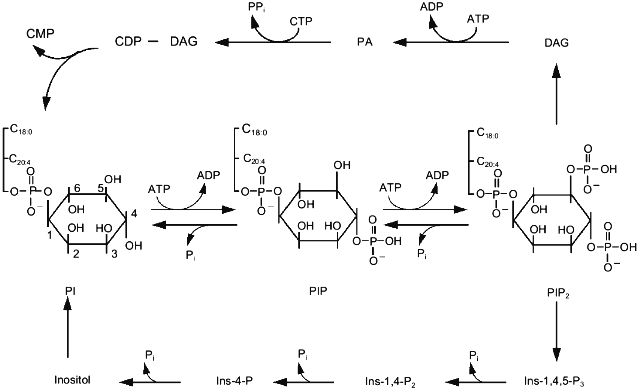
Accumulation of phosphatidylinositol-3,4,5-trisphosphate in the membrane correlates well with actin polymerization. Data on the existence of a specific polyphosphoinositide binding site of gelsolin, villin [24], and profilin [25] suggest that phosphatidylinositol-3,4,5-trisphosphate can participate in the rearrangement of the cytoskeleton.
REGULATION OF PHOSPHOINOSITIDE HYDROLYSIS
Activation of phospholipase C hydrolyzing PI-4,5-P2 leads to formation of diacylglycerol and inositol-1,4,5-trisphosphate. Besides formation of the usual isomer, Ins-1,4,5-P3, the reaction yields some cyclic inositol-1,4,5-trisphosphate (Ins-c1:2,4,5-P3) [26]. The latter is formed when cleavage of the bond between phosphate and glycerol involves hydroxyl group of the inositol ring in the reaction. If the hydroxyl group of water participates in this reaction the phosphodiesterase reaction results in the formation of Ins-1,4,5-P3 [27]. Under physiological condition the proportion of Ins-c1:2,4,5-P3 accumulated during activation of cells by the hormone may reach 30% of the Ins-1,4,5-P3 level [28].
Although PI-4,5-P2 is the main substrate of phospholipase C a possibility of in vivo hydrolysis of other phosphoinositides, PI, and PI-4-P, by phospholipase C was demonstrated. A few isoforms of phospholipase C have been isolated and characterized [29-31]. Each of them can hydrolyze all three phosphoinositides with formation of six products: three non-cyclic (Ins-1-P, Ins-1,4-P2, and Ins-1,4,5-P3) and three cyclic (Ins-c1:2-P, Ins-c1:2,4-P2, and Ins-c1:2,4,5-P3) [32]. Those phosphoinositides, which contain the phosphate group in the third position (PI-3-P, PI-3,4-P2, and PI-1,4,5-P3) are obviously not substrates of phospholipase C [33, 34, 19]. Under physiological conditions phospholipase C hydrolyzes preferentially PI-4-P and PI-4,5-P2, and their hydrolysis requires submicromolar concentrations of Ca2+ (~10-7 M), which is detected in the cytoplasm of resting cells [35].
Subcellular fractionation revealed most phospholipase C is in the cytoplasm, but there are forms of the enzyme that are tightly bound to the plasma membrane [36-38]. Molecular masses of phospholipase C isoforms are within a range of 60-150 kD [29, 30].
Isoforms of phospholipase C are characterized by different primary structure. There is only one homologous domain including 250 amino acids which is located at the same position (250-500 residues) of all isomers [29]. At the same position this domain was also found in retinal phospholipase C of the fruit fly Drosophila [39]. The homology of domain A between isoforms beta, gamma, and delta and phospholipase C of Drosophila was 55-60%, but the homology between these four enzymes and alpha-isoform was only 30-35% [29]. Since domain A is highly conservative and found in all forms of phospholipase C, it probably forms the catalytic site structure of the enzyme. Polypeptide chains of beta-, gamma-, and delta-isoforms and the molecule of phospholipase C of Drosophila contain also one common fragment (B) approximately of the same size as domain A. However, in contrast to domain A, this domain did not have a fixed position.
The molecule of the gamma-isoform of phospholipase C also contains small sequences (about 50 amino acid residues) which are homologous to fragments of such non-receptor tyrosine kinases as src-protein [40]. These sequences are localized between domains A and B. It is suggested that they cause some similarity between the mechanisms of regulation of phospholipase C gamma-isoform and tyrosine kinase src [29]. Besides homology with tyrosine kinase gamma-isoform, phospholipase C has some similarity with GAP protein activating GTPase activity of protein ras p21 [41] and alpha-spectrin [42].
The structure of all phospholipase C isoforms lacks hydrophobic fragments which could provide attachment of the enzyme to phospholipid membrane [29, 30]. Nevertheless, many tissues contain membrane-bound alpha-, beta-, and gamma-isoforms of phospholipase C [30, 38]. After cell activation by epidermal growth factor, platelet-derived growth factor, and nerve growth factor antibodies against corresponding receptors were able to co-precipitate gamma-isoform of phospholipase C. beta-Isoform of phospholipase C was isolated both from repeatedly washed brain membrane fraction and from cytosol [43]. Apparently, various forms of phospholipase C interact in the membrane with proteins (e.g., G-proteins or membrane receptors) rather than with lipids. Attachment of the alpha-isoform of phospholipase C to the membrane occurs after phosphorylation of the enzyme with protein kinase C [38].
The existence of a large number of phospholipase C isoforms can be explained by the involvement of this enzyme into signal transduction of many different agonists [44]. Hormones acting at receptors coupled to G-proteins (mainly Gq-proteins) activate the beta-isoform of phospholipase C, the gamma-isoform responds to the effects of growth factors [31] (Fig. 2).
More than 30 receptors related to a family of membrane proteins with seven transmembrane domains act via stimulation of the beta-isoform of phospholipase C. These are alpha1-adrenergic, M1-, M3-, and M5-muscarinic cholinergic, P2t- and P2y-purinergic, 5HT1c-serotoninergic, V1a and V1b vasopressin, H1-histamine receptors, and also receptors of gonadoliberin, cholecystokinin, oxytocin, angiotensin II, thrombin, bradykinin, vasoactive intestinal peptide, thromboxanes, endothelin, platelet aggregating factor, and many other hormones [45].Fig. 2. Mechanisms of action of hormones and growth factors on the cell via the activation of phospholipase C (PLC) specific with respect to PIP2. G, G-protein; P, inorganic phosphate; other abbreviations are the same as in Fig. 1.
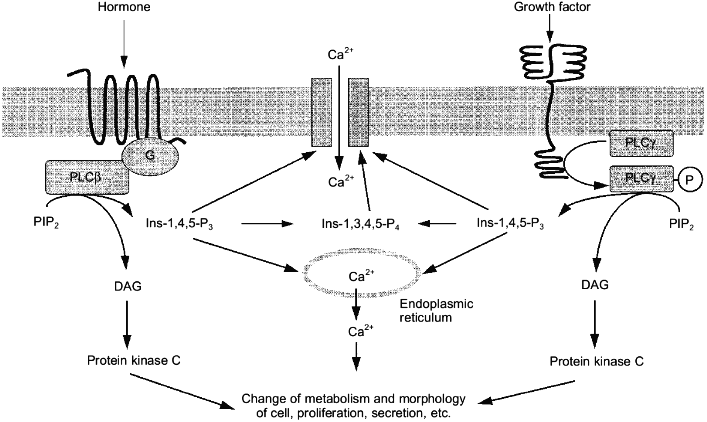
Mixing of the alpha-subunit of the Gq-protein and the beta1-isoform of phospholipase C isolated from brain [46, 47] resulted in a 10-20-fold increase of the rate of PI-4,5-P2 hydrolysis. Gq-Protein did not influence the gamma- and delta-isoforms [47]. Since homogenous preparations of phospholipase and Gq-protein were used in the experiments and the activation was observed at their equimolar ration the formation of an active complex between the enzyme and the GTP-binding subunit of the Gq-protein was postulated.
Besides Gq-protein, alpha-subunits of G11-, G14-, and G16-protein can activate the beta-isoform of phospholipase C [48-50]. Pertussis toxin-sensitive G-proteins (Gi and Go) can also interact with this isoform of phospholipase C [51]. It was convincingly demonstrated that phospholipase C can bind both alpha-subunit and beta-gamma-dimer of G-proteins. Binding of both alpha- and beta-gamma-subunits occurs independently at various sites on the enzyme. beta-gamma-Dimer of G-proteins as well as the alpha-subunit activate the beta-isoform of phospholipase C [52].
Low molecular weight G-proteins with molecular mass of 21 kD encoded by genes of the ras group (Ha-ras, Ki-ras, and N-ras) are considered as possible regulators of phospholipase C. Transformation of 3T3 fibroblasts by oncogene ras increased stimulation of phospholipase C by muscarinic cholinergic agonists [53], but attenuated activation of adenylate cyclase by catecholamines acting at beta-adrenoreceptors. Fibroblasts transformed by any of three oncogenes are characterized by increased concentration of diacylglycerol [54] and also by increased level of inositol phosphates, suggesting an increased role of PI-4,5-P2 hydrolysis.
In platelets and human erythroleukemia cells ras-like membrane GTP-binding protein rap1-b with molecular mass of 22 kD participates in transduction of hormonal signal on phospholipase C [55]. This protein is one of the main substrates of cAMP-dependent protein kinase in these cells [56]. It is suggested that in platelets and erythroleukemia cells the oligomeric protein complex receptor--protein--rap1-b--phospholipase--alpha-subunit of heterotrimeric G-protein is formed during activation of phospholipase C [55]. In the case of thrombin action on cells this process involves the alpha-subunit of the Gi2-protein [57]. Adenylate cyclase activators (e.g., prostacyclin) increase cAMP level, leading to phosphorylation of protein rap1-b. Some evidence exists that rap1-b causes effects of thrombin and other agonists on the cytoskeleton [58].
Receptors of growth factors differ from hormonal receptors coupled to G-protein by structure. They only once span the membrane and contain a cytoplasmic domain possessing tyrosine kinase activity (Fig. 2). Activation of PI-4,5-P2 by epidermal and platelet-derived growth factors is accompanied by incorporation of phosphate into tyrosine and serine amino acid residues of the gamma-isoform of phospholipase C [59]. Phosphorylation was observed not only in intact cells, but also when these homogenous receptors of growth factors were added to homogenous isoform of the enzyme [59, 60]. Binding of platelet derived or epidermal growth factors to their receptors causes the formation of complexes between these receptors and phospholipase C [59, 61]. Mutations leading to the disappearance of tyrosine kinase activity in receptors resulted in a lack of activation of phosphoinositide metabolism by growth factors [62, 63]. Tyrosine kinase inhibitors abolished activation of phospholipase C by growth factors [64]. Thus, all these data suggested that the activation of the gamma-isoform of phospholipase C by growth factors occurs as a result of phosphorylation of its tyrosine residue.
Phospholipase C is a Ca2+-binding protein possessing the EF-domain typical of other Ca2+-binding proteins; it provides high affinity Ca2+ binding [65]. At millimolar concentrations of Ca2+, the rates of hydrolysis of phosphatidylinositol, phosphatidylinositol-4-phosphate, and phosphatidylinositol-4,5-bisphosphate are comparable. However, at nearly physiological Ca2+ concentrations (1-10 µM) the efficacy of PI hydrolysis sharply decreased and PI-4-P and PI-4,5-P2 become preferential substrates [29].
Decrease of phospholipase C activity can be achieved by lowering the cytoplasmic Ca2+ concentration. Perhaps, this is a way for inhibition of phosphoinositide metabolism by dopamine mediated in pituitary lactotrophic cells via D2-receptors. It is suggested that dopamine activates potassium channels, this resulting in hyperpolarization of the membrane and inhibition of Ca2+ influx via potential-dependent calcium channels. Decrease of Ca2+ level in the cytoplasm causes a decrease in inositol phosphate formation. In platelets all activators of adenylate cyclase (prostacyclin, prostaglandin E1, etc.) decrease phosphoinositide metabolism [6]. Similar effects of adenylate cyclase activators was observed in smooth muscle cells. In these cells phosphoinositide metabolism is also blocked by an increase in cGMP level induced by nitroprusside or atrial natriuretic factor. These effects of cAMP and cGMP can be explained by blockade of Ca2+ influx into the cell [45].
Since Ca2+ increases catalytic activity of practically all isoforms of phospholipase C, increase of Ca2+ level in cytoplasm to 10-6-10-5 M caused activation of phosphoinositide metabolism. Perhaps this represent a mechanism for formation of diacylglycerol and inositol phosphates during stimulation of lipid peroxidation, membrane damage, the effect of Ca-ionophores, and the mechanical stretching of cells.
METABOLISM OF INOSITOL PHOSPHATES
Two enzymes catalyze conversions of Ins-1,4,5-P3 in cells. 5-Phosphatase cleaves the phosphate group at position 5. 3-Kinase catalyzes ATP-dependent phosphorylation of inositol-trisphosphate at position 3 (Fig. 3). There are both membrane-bound and soluble isoforms of 5-phosphatase [66, 67]. Soluble 5-phosphatase from platelet cytosol can cleave the phosphate group from three phosphosugars: Ins-1,4,5-P3, Ins-1,3,4,5-P4, and Ins-c1:2,4,5-P3 [68]. The enzyme has one order of magnitude higher affinity for Ins-1,3,4,5-P4 (Km ~ 1 µM) than to Ins-1,4,5-P3, so at comparable concentrations of these substrates it preferentially hydrolyzes Ins-1,3,4,5-P4 [66]. However, Vmax for Ins-1,4,5-P3 hydrolysis is 3-fold higher than that for Ins-1,3,4,5-P4 hydrolysis. The hydrolysis of the third substrate of 5-phosphatase, Ins-c1:2,4,5-P3, occurs more slowly because Vmax of this reaction is one order of magnitude less than Vmax for the first two substrates, and Km is ~150 µM.
Theoretically, the product of the 5-phosphatase reaction, Ins-1,4-P2, can be cleaved by two phosphatases hydrolyzing phosphoester bonds at positions 1 or 4; however, in reality it is further metabolized with formation of Ins-4-P [69-71], i.e., 1-phosphatase catalyzes hydrolysis of Ins-1,4-P2 (Fig. 3). Marked hydrolysis of Ins-1,4-P2 catalyzed by 4-phosphatase was observed only at high concentrations of the substrate (10-4-10-3 M) [72, 73]. 1-Phosphatase is localized in cytoplasm, but a small portion of the enzyme can be membrane-bound [66]. Besides Ins-1,4-P2 1-phosphatase catalyzes hydrolysis of Ins-1,3,4-P3. The affinity of this enzyme to Ins-1,4-P2 and Ins-1,3,4-P3 is 5 and 20 µM, respectively [74, 75]. Millimolar concentrations of Li+ inhibit noncompetitively the 1-phosphatase activity [66].Fig. 3. Metabolism of inositol phosphates in the cell. Ins-1,3,4,5-P4, inositol tetrakisphosphate; other abbreviations are given in the legend to Fig. 1.
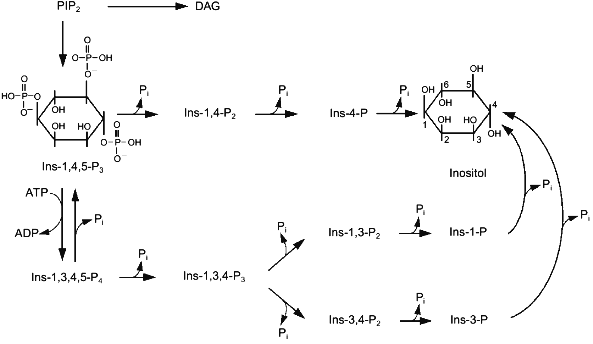
Inositol-4-phosphate is the last product in the alpha pathway of breakdown of phosphoinositides to inositol. It is hydrolyzed by a non-specific inositol monophosphate phosphatase (Fig. 3). This enzyme catalyzes hydrolysis of other isomers of inositol monophosphate formed during metabolism. Inositol monophosphate phosphatase is a dimer formed by two subunits with molecular mass of 29 kD [76]. Like some other enzymes of inositol phosphate metabolism, this phosphatase is inhibited by Li+.
The second route of Ins-1,4,5-P3 metabolism begins with a phosphorylation reaction catalyzed by the 3-kinase (Fig. 3). This results in formation of Ins-1,3,4,5-P4, which can be converted into Ins-1,3,4-P3 by 5-phosphatase or dephosphorylated to Ins-1,4,5-P3 by inositol-1,3,4,5-tetrakisphosphate 3-phosphomonoesterase [77, 78]. 3-Kinase is highly specific with respect to Ins-1,4,5-P3, Km for the enzyme from different types of cells varies from 0.2 to 1.5 µM [66]. Increase of Ca2+ (from 10-7 M to micromolar concentrations) stimulates the activity of the 3-kinase in a calmodulin-dependent manner [79-81]. Thus, the increase of intracellular Ca2+ concentrations induced by Ins-1,4,5-P3 promotes its subsequent conversion into Ins-1,3,4,5-P4. Formation of Ins-1,3,4,5-P4 and its metabolite Ins-1,3,4-P3 was actually found in cells under the influence of hormones increasing the concentration of cytoplasmic Ca2+ ([Ca2+]cyt) [82]. The importance of the second pathway of Ins-1,4,5-P3 metabolism is not limited only by removal of the second messenger, i.e., to quenching of the signal caused by the increase in [Ca2+]cyt. This reaction with Ins-1,3,4-P3 begins formation of other higher inositol phosphates which probably have important biological functions [82-85]. Ins-1,3,4,5-P4 also regulates [Ca2+]cyt. This compound activates a Ca2+ current from the external medium into cells [86] and also to intracellular stores [87]. Taking into consideration data on the conversion of Ins-1,3,4,5-P4 into Ins-1,4,5-P3 catalyzed by inositol tetrakisphosphate 3-phosphomonoesterase, it may be suggested that Ins-1,3,4,5-P4 is a buffer which increases resources of Ins-1,4,5-P3, a Ca2+-mobilizing second messenger.
OSCILLATION OF Ca2+ IONS
Structures providing Ca2+ influx into the cytoplasm via a gradient of its concentration (Ca2+-channels) and systems of active transport of Ca2+ against its concentration gradient utilizing either the energy of ATP (Ca2+-pump) or gradients of other ions (Na/Ca carrier) operate in cell membranes [45]. Combined functioning of systems of passive or active Ca2+ transport via cytoplasmic and intracellular membranes provides so-called "transient" increase of Ca2+ concentrations. This implies normalization of cytoplasmic Ca2+ to its basal level irrespectively to whether signal inducing Ca2+ influx into a cell still acts. Functioning of different Ca2+-channels in the cell connected by positive and negative feedback, interaction of these channels with systems of active Ca2+ transport, and Ca2+-binding proteins leads to oscillation of cytoplasmic concentrations of Ca2+. The frequency of this oscillation depends on the products of the phosphoinositide hydrolysis and is increased by various extracellular stimuli (hormones, growth factors, and mechanical irritation of the cell).
The receptor of Ins-1,4,5-P3 is a homotetramer; its subunits form a non-specific pore for cations [88]. The primary structure of this protein is close to that of the ryanodine receptor, but it has a low similarity with the structure of the potential-dependent Ca2+-channel. The receptor of Ins-1,4,5-P3 is a channel former which is a Ca2+-binding protein. Low concentrations of Ca2+ activate whereas high concentrations inhibit it. Ins-1,4,5-P3 binds to its receptor in a cooperative manner, resulting in desensitization, i.e., decrease of receptor sensitivity to its agonist. This channel also binds ATP and can be phosphorylated by cAMP-dependent protein kinase [89, 90].
The ryanodine receptor is expressed in excitatory cells and can function in membranes of endoplasmic reticulum concordantly to Ins-1,4,5-P3 receptor. As the latter, ryanodine is a tetramer forming non-selective cation pore through which Ca2+ can enter the cytoplasm. This channel is activated by micromolar concentrations and inhibited by millimolar concentrations of Ca2+ [91]. Caffeine, the methylxanthine of plant origin, is a well known ligand for this receptor. Cyclic ADP-ribose is apparently the endogenous ligand [92]. A local increase of Ca2+ concentration induced by the influx of the external cation is also able to activate the ryanodine Ca2+-channel. Interaction between Ca2+-channels of the outer and inner membranes, Ca2+-pumps, and also Ca2+-binding proteins localized both in membranes and in the cytoplasm leads to periodical fluctuations of Ca2+ in the cytoplasm [45, 93].
In non-excitatory tissues Ins-1,4,5-P3 is the main trigger of calcium oscillations. It is formed from PIP2 on the activation of phospholipase C by hormones or growth factors. Within decades of seconds Ins-1,4,5-P3 is able to diffuse from the plasma membrane where it is formed to the membrane of the endoplasmic reticulum. Quantity and concentration of forming Ins-1,4,5-P3 are high enough to occupy all molecules of corresponding receptors; however, Ca2+ efflux occurs only at so called "hot" sites. These sites of transient localization in cells appearing due to high local concentration of Ca2+, Ins-1,4,5-P3 or its receptor. According to modern concepts [94], Ca2+ released at hot site into the cytoplasm diffuses along the reticulum and increases receptor sensitivity to Ins-1,4,5-P3 and also facilitates channel opening, thus causing a shift of the front of a Ca2+ wave. Local augmentation of Ca2+ concentration at this membrane site leads to inactivation of the Ca2+-channel that causes quenching of the hot point, whereas Ca2+ diffusion generates new points of Ca2+ release from the reticulum. Ca2+-pumps transporting Ca2+ into the reticulum or intracellular space are also involved into quenching of hot points.
In excitatory tissues the main Ca2+ entry into cells occurs via potential-dependent Ca2+-channels, which are functionally coupled to ryanodine receptors [95]. At the coupling point of these two types of channels Ca2+ release leads to spreading of a Ca2+ wave of ryanodine channel excitation along the membrane of the reticulum. Decrease of Ca2+ level due to exhaustion of the reticulum and Ca2+ removal from the cytoplasm by the Ca2+ pump occurs after the front of the Ca2+ wave. In excitatory cells Ins-1,4,5-P3, cyclic ADP-ribose, or caffeine can increase the frequency of Ca2+ oscillations.
In many cell types the wave of calcium oscillation propagates from the cellular nucleus and can acquire forms of spheres or complex helices. In some tissues (heart, brain, and endothelium) Ca2+ oscillations appearing in a single cell can induce in neighboring cells Ca2+ oscillations of the same frequency as in the cell initiating this process. Apparently, in these tissues the Ca2+ wave can be propagated via intercellular contacts possessing high ion conductivity [45].
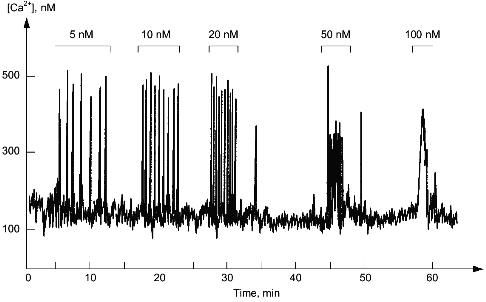
In the cytoplasm of a single cell, hormones and growth factors have practically no influence on the amplitude of the increase of the Ca2+ concentration but increase the frequency of its fluctuations (Fig. 4). Usually the Ca2+ level in the cytoplasm changes from 10-7 to 5·10-7 M, whereas the frequency varies from one oscillation per minute to a few oscillations per second. Ca2+-Dependent effects of hormones and growth factors strictly depend on the frequency of the fluctuations of cytoplasmic Ca2+. Apparently, this can be explained by increased probability of saturation of Ca2+-binding protein with calcium during the increase of frequency of oscillations. Dissociations of Ca2+ from high affinity sites of Ca2+-binding proteins accept this "frequency" information and transform it into slow developing (within minutes or hours) change of metabolism, morphology, or functional state of the cell.
Fig. 4. Fluctuations of Ca2+ level in the cytoplasm of a single smooth muscle cell. The upper part shows concentrations of endothelin-1, activator of phosphoinositide metabolism in these cells added to the external medium.
REFERENCES
1.Hokin, M. R., and Hokin, L. E. (1953) J. Biol.
Chem., 203, 967-977.
2.Hokin, L. E., and Hokin, M. R. (1958) J. Biol.
Chem., 233, 805-810.
3.Michell, R. H. (1975) Biochim. Biophys.
Acta, 415, 81-147.
4.Berridge, M. J., Dawson, R. M., Downes, C. P.,
Heslop, J. P., and Irvine, R. F. (1983) Biochem. J.,
212, 473-482.
5.Streb, H., Irvine, R. F., Berridge, M. J., and
Schultz, I. (1983) Nature, 306, 67-69.
6.Irvine, R. F., Lethcher, A. J., Lander, D. J., and
Downes, C. P. (1984) Biochem. J., 232, 237-243.
7.Burgess, G. M., McKinney, J. S., Irvine, R. F., and
Rutney, J. W., Jr. (1985) Biochem. J., 232, 237-243.
8.Batty, I. R., Nahorski, S. R., and Irvine, R. F.
(1985) Biochem. J., 232, 211-215.
9.Balla, T., Baukal, A. J., Guillemette, G., and
Catt, K. J. (1988) J. Biol. Chem., 263, 4083-4091.
10.Hansen, C. A., vom Dahl, S., Huddell, B., and
Williamson, J. R. (1988) FEBS Lett., 236, 53-56.
11.Tilly, B. C., van Paridon, P. A., Verlaan, I.,
Wirtz, K. W. A., de Laat, S. W., and Molenaar, W. H. (1987)
Biochem. J., 244, 129-135.
12.Szwergold, B. S., Graham, R. A., and Brown, T. R.
(1987) Biochem. Biophys. Res. Commun., 149, 874-881.
13.Vallejo, M., Jackson, T., Lightman, S., and
Hanley, M. R. (1987) Nature, 330, 656-658.
14.Kirk, C. J., Barker, C. J., Craxton, A.,
Estrada-Garcia, T., Evans, W. H., Parry, J. B., Potter, B. L., Shears,
S. B., Wong, N.-S., and Michell, R. H. (1989) Arch. Pharmacol.,
78, 7-8.
15.Majerus, P. W., Connolly, T. M., Deckmyn, H.,
Ross, T. S., Bross, T. E., Ishii, H., Bansal, V. E., and Wilson, D. B.
(1986) Science, 234, 1519-1524.
16.Hokin, L. E. (1985) Ann. Rev. Biochem.,
54, 205-235.
17.Carpenter, C. L., Duckworth, B. C., Auger, K. R.,
Cohen, B., Schaffhausen, B. S., and Cantley, L. C. (1990) J. Biol.
Chem., 265, 19704-19711.
18.Auger, K. R., Serunian, L. A., Soltoff, S. P.,
Libby, P., and Cantley, L. C. (1989) Cell, 57,
167-175.
19.Kucera, G. L., and Rittenhouse, R. E. (1990)
J. Biol. Chem., 265, 5345-5348.
20.Nakanishi, H., Brewer, K. A., and Exton, J. H.
(1993) J. Biol. Chem., 268, 13-16.
21.Nishizuka, Y. (1984) Nature, 308,
693-698.
22.Hug, H., and Sarre, T. F. (1993) Biochem.
J., 291, 329-343.
23.Divecha, N., and Irvine, R. F. (1985)
Cell, 80, 269-278.
24.Janmey, P. A., Lamb, J., Allen, P. G., and
Matsudaira, P. T. (1992) J. Biol. Chem., 267,
11818-11823.
25.Janmey, P. A., and Stossel, T. P. (1987)
Nature, 325, 362-364.
26.Wilson, D. B., Connolly, T. M., Bross, T. E.,
Majerus, P. W., Sherman, W. R., Tyler, A. N., Rubin, L. J., and Brown,
J. E. (1985) J. Biol. Chem., 260, 13496-13501.
27.Dawson, R. M., Freinkel, N., Jungalwala, F. B.,
and Clarke, N. (1971) Biochem. J., 122, 605-607.
28.Sekar, M. C., Dixon, F. J., and Hokin, L. E.
(1987) J. Biol. Chem., 262, 340-344.
29.Crooke, S. T., and Bennet, C. F. (1989) Cell
Calcium, 10, 309-323.
30.Rhee, S. G., Suh, P. G., Rye, S. H., and Lee, S.
Y. (1989) Science, 244, 546-550.
31.Rhee, S. G., and Choi, K. D. (1992) J. Biol.
Chem., 267, 12393-12396.
32.Majerus, P. W., Connoly, T. M., Bansal, V. S.,
Inhorn, R. C., Ross, T. S., and Lips, D. L. (1988) J. Biol.
Chem., 263, 3051-3054.
33.Whitman, M., Downes, C. P., Keeler, M., Keller,
T., and Cantley, L. (1988) Nature, 332, 644-646.
34.Traynor-Kaplan, A. E., Harris, A. L., Thompson,
B. L., and Sklar, L. A. (1988) Nature, 334, 353-356.
35.Culty, M., Davidson, M. L., and Haslam, R. J.
(1988) Eur. J. Biochem., 171, 523-533.
36.Morris, A. J., Waldo, G. L., Downes, C. P., and
Harden, T. K. (1990) J. Biol. Chem., 265,
13501-13507.
37.Morris, A., Waldo, G. L., Downes, C. P., and
Harden, T. K. (1990) J. Biol. Chem., 265,
13508-13514.
38.Bennett, C. F., and Crooke, S. T. (1987) J.
Biol. Chem., 262, 13789-13797.
39.Bloomquist, B. T., Shortridge, R. D., Schneuwly,
S., Perdew, M., Montell, C., Steller, H., Rubin, G., and Pak, W. L.
(1988) Cell, 54, 723-733.
40.Stahl, M. L., Ferenz, C. R., Kelleher, K. L.,
Kriz, R. W., and Knopf, J. L. (1988) Nature, 332,
269-272.
41.Vogel, U. S., Dixon, R. A. F., Schaber, M. D.,
Diehl, R. E., Marshall, M. S., Scolnick, E. M., Sigal, I. S., and
Gibbs, J. B. G. (1988) Nature, 335, 90-93.
42.Lehto, V. P., Vasenius, V.-M., Salven, P., and
Saraste, M. (1988) Nature, 334, 388.
43.Katan, M., and Parker, P. J. (1987) Eur. J.
Biochem., 168, 413-418.
44.Bennett, C. F., Balcarek, J. M., Varrichio, A.,
and Crooke, S. T. (1988) Nature, 334, 268-271.
45.Avdonin, P. V., and Tkachuk, V. A. (1994)
Receptors and Intracellular Calcium [in Russian], Nauka,
Moscow.
46.Taylor, S. J., and Exton, J. H. (1991) FEBS
Lett., 286, 214-216.
47.Taylor, S. J., Chae, H. Z., Ree, S. G., and
Exton, J. H. (1991) Nature, 350, 516-518.
48. Lee, C. H., Park, D., Wu, D., Rhee, S. G., and
Simon, M. I. (1992) J. Biol. Chem., 267,
16044-16047.
49.Smrcka, A. V., Hepler, J. R., Brown, K. O., and
Sternweis, P. C. (1991) Science, 251, 804-807.
50.Taylor, C. W., and Marshall, I. C. B. (1992)
Trends Biochem. Sci., 17, 502-506.
51.Sternweis, P. C., and Smrcka, A. V. (1992)
Trends Biochem. Sci., 17, 502-506.
52.Clapham, D. E., and Neer, E. J. (1993)
Nature, 365, 403-406.
53.Chiarugi, V., Porciatti, F., Pasquali, F., and
Bruni, P. (1985) Biochem. Biophys. Res. Commun., 132,
900-907.
54.Fleischman, L. F., Chahwala, S. B., and Cantley,
L. (1986) Science, 231, 407-410.
55.Lapetina, E. G. (1990) FEBS Lett.,
268, 400-404.
56.White, T. E., Lacal, J. C., Reep, B., Fischer, T.
H., Lapetina, E. G., and White II, G. S. (1990) Proc. Natl. Acad.
Sci. USA, 87, 785-762.
57.Lazarowski, E. R., and Lapetina, E. G. (1990)
Arch. Biochem. Biophys., 276, 265-269.
58. Fischer, T. H., Gatling, M. N., Lacal, J.-C.,
and White II, G. C. (1990) J. Biol. Chem., 265,
19405-19408.
59.Meisenhelder, J., Suh, P.-G., Rhee, S. G., and
Hunter, T. (1989) Cell, 57, 1109-1122.
60.Nishibe, S., Wahl, M. I., Rhee, S. G., and
Carpenter, G. (1989) J. Biol. Chem., 264,
10335-10338.
61. Margolis, B., Li, N., Koch, A., Mohammadi, M.,
Hurwitz, D. R., Zilberstein, A., Ullrich, A., Pawson, T., and
Schessinger, J. (1990) EMBO J., 9, 4375-4380.
62. Molenaar, W. H., Bierman, A. J., Tilly, B. C.,
Verlaan, I., Defize, L. H. K., Honegger, A. M., Ulrich, A., and
Schlessinger, J. (1988) EMBO J., 7, 707-710.
63.Keating, M. T., Escobedo, J. A., and Williams, L.
T. (1988) J. Biol. Chem., 263, 12805-12808.
64.Margolis, B., Rhee, S. G., Felder, S., Mernic,
M., Lyall, R., Levitzki, A., Ullrich, A., Zilberstein, A., and
Schlessinger, J. (1989) Cell, 57, 1101-1107.
65.Bairoch, A., and Cox, J. A. (1990) FEBS
Lett., 269, 454-456.
66.Shears, S. B. (1989) Biochem. J.,
260, 313-324.
67.Hansen, C. A., Johanson, R. A., Williamson, M.
T., and Williamson, J. R. (1987) J. Biol. Chem., 262,
17319-17326.
68. Connolly, T. M., Bross, T. E., and Majerus, P.
W. (1985) J. Biol. Chem., 260, 7868-7874.
69.Inhorm, R. C., Bansal, V. S., and Majerus, P. W.
(1987) Proc. Natl. Acad. Sci. USA, 84, 2170-2174.
70.Morgan, R. O., Chang, J. P., and Catt, K. S.
(1987) J. Biol. Chem., 262, 1166-1171.
71.Dillon, S. B., Murray, J. J., Verhese, M. W., and
Snyderman, R. (1987) J. Biol. Chem., 262,
11546-11552.
72.Hughes, A. R., and Putney, J. W., Jr. (1989)
J. Biol. Chem., 264, 9400-9407.
73.Ackermann, K. E., Gish, B. J., Honchar, M. P.,
and Sherman, W. R. (1987) Biochem. J., 242, 517-524.
74.Inhorn, R. C., and Majerus, P. W. (1987) J.
Biol. Chem., 262, 15946-15952.
75.Inhorn, R. C., and Majerus, P. W. (1988) J.
Biol. Chem., 263, 14559-14565.
76.Gee, N. S., Ragan, C. A., Watling, C. J., Aspley,
S., Jackson, R. G., Reid, G. G., Gani, D., and Shute, J. K. (1988)
Biochem. J., 249, 883-889.
77.Doughney, C., McPherson, M. A., and Dormer, R. L.
(1988) Biochem. J., 251, 927-929.
78.Shears, S. B. (1990) Cell. Signal.,
2, 191-195.
79.Morris, A. P., Gallacher, D. V., Irvine, R. F.,
and Petersen, O. H. (1987) Nature, 330, 653-655.
80.Daniel, J. L., Dangelmaier, C. A., and Smith, J.
B. (1988) Biochem. J., 253, 789-794.
81. Biden, T. J., and Wollheim, C. B. (1986) J.
Biol. Chem., 261, 11931-11934.
82.Berridge, M. J., and Irvine, R. F. (1989)
Nature, 341, 197-205.
83.Downes, C. P. (1988) Trends Neurosci.,
11, 336-338.
84.Fink, L. A., and Kaszmarek, L. K. (1988)
Trends Neurosci., 11, 338-339.
85.Joseph, S. K., and Williamson, J. R. (1989)
Arch. Biochem. Biophys., 273, 1-15.
86.Irvine, R. F., and Moor, R. M. (1986) Biochem.
J., 240, 917-920.
87.Hill, T. D., Dean, N. M., and Boynton, A. L.
(1988) Science, 242, 1176-1178.
88.Mikoshiba, K. (1993) Trends Pharmacol.
Sci., 14, 86-89.
89.Parys, J. B., Sernett, S. W., De Lisle, S.,
Shyder, P. M., Welsh, M. J., and Campbell, K. P. (1992) J. Biol.
Chem., 267, 18776-18782.
90. Finch, E. A., and Goldin, S. M. (1994)
Science, 265, 813-815.
91.Ehrlich, B. E., Kaftan, E., Bezprozvannaya, S.,
and Bezprozvannaya, A. (1994) Trends Pharmacol. Sci.,
15, 145-149.
92.Thorn, P., Gerasimenko, O., and Petersen, O. H.
(1994) EMBO J., 13, 2038-2043.
93.Clapham, D. E. (1995) Cell, 80,
259-268.
94.Atri, A., Amundson, J., Clapham, D. E., and
Sneyf, J. (1993) Biophys. J., 65, 1727-1739.
95.McPherson, P. S., and Cambell, K. P. (1993) J.
Biol. Chem., 268, 13765-13768.