REVIEW: 2-Arachidonoyl-glycerol as an "Endocannabinoid": Limelight for a Formerly Neglected Metabolite
V. Di Marzo
Istituto per la Chimica di Interesse Biologico, C.N.R., Via Toiano 6, 80072, Arco Felice, Napoli, Italy; fax: 39 (81) 804-1770; E-mail: vdm@trinc.icmib.na.cnr.it
Submitted May 28, 1997
Previously believed to simply be an intermediate in tri- and diglyceride metabolism or an alternative precursor for arachidonic acid, 2-arachidonoyl-glycerol has lately attracted renewed interest from lipid biochemists and pharmacologists. This is due to the finding of its cannabimimetic activity. In the present article recent landmarks that have led to the proposition of a role of this monoglyceride as an "endocannabinoid", starting from its newly discovered pharmacological properties in both central and peripheral tissues and ending with studies on the possible biosynthetic pathways for its formation are reviewed. Also considered are possible interactions with another arachidonic acid-derived endogenous cannabinoid, anandamide.
KEY WORDS: cannabinoids, anandamide, arachidonic acid, eicosanoids, cannabinoid receptors, oxylipins
Abbreviations: AA) arachidonic acid; AA-CoA) arachidonoyl-coenzyme A; AArG) diglycerides bearing AA in sn-2 position; ANA) anandamide (ethanolamide of AA); 2-AG) 2-arachidonoyl-glycerol; N-ArPE) N-arachidonoyl-phosphatidylethanolamine; AAPC) 1,2-sn-diarachidonoyl-phosphatidylcholine; CB) cannabinoid receptor; DAG) diacylglycerol; MAG) monoacylglycerol; FAAH) fatty acid amide hydrolase; PEA) palmitylethanolamide; PLÀ2, PLC, PLD) phospholipase À2, C, and D, correspondingly; PC) phosphatidylcholine; PKC) protein kinase C; PUFA) polyunsaturated fatty acids; THC) Delta9-tetrahydrocannabinol.
Research started in the late 1970's and continued throughout the 1980's
and 90's has led to the conclusive recognition of the importance of
phospholipid-derived molecules as intra- and extra-cellular signals in
animal cells [1]. In this context, particularly
significant appears to be the role played by arachidonic acid (AA), as
well as other polyunsaturated fatty acids (PUFAs), either as
precursors, once released from membrane phosphoglycerides by the action
of various phospholipases, of a plethora of bioactive oxidation
products, the oxylipins [2], or as the major
components esterified at the sn-2 position of both phospholipid
and phospholipid-derived chemical signals such as the phosphoinositides
(PIs), the diacylglycerols (DAGs), the (lyso)phosphatidic acids, and
the platelet activating factor. Among the possible derivatives of the
non-oxidative metabolism of AA, the monoglyceride
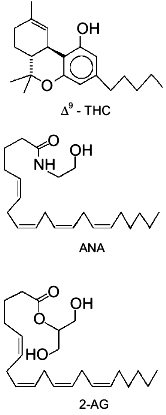
2-arachidonoyl-glycerol (2-AG, Fig. 1) has been
often described to occur together with other components of the above
mentioned families of lipids in both resting and stimulated cells [3, 4]. However, the absence in the
literature of data describing any kind of pharmacological action for
this compound, and its lack of activity on DAG, the major target in the
cell, i.e., protein kinase C (PKC), led to consider 2-AG simply as one
of the possible inactive degradation products of DAGs [3]. The subsequent finding of sn-1 specific
DAG-lipase enzymes (for example, see [5]) seemed to
corroborate this hypothesis. Another possibility was that 2-AG derived
from the hydrolysis of DAGs might be used as a precursor for the
formation of AA in cells where the stimulus-induced release of this
PUFA occurs upon activation of phospholipase C (PLC) and subsequent
breakdown of PIs or phosphatidylcholine (PC). Indeed, before the
finding that receptors can directly activate phospholipase A2
(PLA2) through GTP-binding (G) proteins, DAG-lipase
mediated release of AA from DAGs derived from the catalytic action of
PLC was thought to be the only source of agonist-induced AA release
([6-8] and for a recent review
[9]). However, also due to the lack of selective
inhibitors for sn-1- and sn-2-DAG-lipases, to date only
few studies [5, 10-12] have managed to clarify the issue of whether AA
is released from DAGs directly through the action of a
sn-2-DAG-lipase, or via the monoacylglycerol
(MAG)-lipase-catalyzed hydrolysis of sn-1-DAG-lipase-derived
2-AG. Therefore, the role of the latter compound as a metabolic source
of AA is yet to be fully assessed.
In summary, until very recently, evidence suggesting for 2-AG a physiological role other than that of metabolic intermediate and/or by-product was limited to the finding of: 1) a positive modulatory action on Na+/K+-ATPase activity [13], and 2) a possible alternative biosynthetic pathway for the monoglyceride in rat brain, starting from PIs and through the sequential action of a PI-selective phospholipase A1 and a lysoPI-specific PLC [14]. The recent breakthroughs which have suggested for 2-AG a role as endogenous ligand of cannabinoid receptors, thus suddenly bringing this glyceride to a foreground position in the field of bioactive lipid research, are reviewed in the present article.Fig. 1. Chemical structures of Cannabis sativa major active principle (-)Delta9-tetrahydrocannabinol (Delta9-THC), anandamide (ANA), and 2-arachidonoyl-glycerol (2-AG). The structures of the latter two compounds are drawn in a way to stress their similarity with Delta9-THC chemical structure.
ENDOCANNABINOIDS AND THE ENDOGENOUS CANNABINOID SYSTEM
Almost to underline further the importance of PUFAs in chemical signalling, in 1992 the discovery came [15] that the long-looked for endogenous ligand of mammalian brain cannabinoid (CB1) receptors was another derivative of the non-oxidative metabolism of AA, i.e., arachidonylethanolamide, the amide of AA with ethanolamine (Fig. 1). Due to its possible function as an endogenous cannabimimetic substance, originally supported by its capability to selectively bind with high affinity to membrane preparations containing CB1 receptors [15], this compound was named anandamide, from the Sanskrit word "ananda" (for "bliss"). That anandamide (ANA) might be a likely candidate for the role of agonist at cannabinoid receptors was immediately clear from the initial data on its pharmacological and behavioral properties, which were almost identical to those of Cannabis major active principle, (-)Delta9-tetrahydrocannabinol (THC) [16-20]. Further studies (reviewed in [21, 22]) showed that ANA shared with THC most of its pharmacological actions, both in vivo and in vitro, in the CNS as well as in some peripheral tissues and cells. However, some subtle differences in the actions of the two endogenous and xenobiotic cannabimimetic metabolites were also noticed, which led to suggest that, in some cases, ANA may act as a partial agonist at CB1 receptors, thus implying the existence of other "endocannabinoids", and that, in any event, the physiological role of ANA could not be simply inferred from the pharmacological data previously reported for plant and synthetic cannabinoids (reviewed in [23]). On the other hand, the hypothesis that ANA might be a novel neuronal mediator was strongly supported by the finding, in intact rat central neurons, of a molecular mechanism by which the cannabimimetic metabolite was: a) biosynthesized in a Ca2+-dependent fashion upon treatment with membrane depolarizing stimuli, and b) inactivated via a re-uptake mechanism and through the hydrolysis of the amide bond [24]. It was found that ANA, in agreement with a pathway previously reported only for saturated and monounsaturated acylethanolamides [25], could be released, in the CNS, from a preformed phospholipid precursor, N-arachidonoyl-phosphatidylethanolamine (N-ArPE), through the action of a phospholipase D-like enzyme [24, 26, 27]. N-ArPE, in turn, as previously described for other N-acyl-phosphatidylethanolamines, was shown to be derived from the Ca2+-dependent enzymatic trans-acylation of the N-position of phosphatidylethanolamine, with 1,2-sn-diarachidonoyl-phosphatidylcholine (AAPC) serving as a possible arachidonate donor [26-29] (Fig. 3). This pathway was suggested to underlie ANA formation also in rat testes [30], murine leukocytes [31, 32], and sea urchin ovaries [33] and is probably responsible for the stimulus-induced biosynthesis of the "endocannabinoid" in most cells. However, the enzymatic, energy-free condensation of AA with millimolar concentrations of ethanolamine was also found to lead to ANA formation in cell-free homogenates, but not in intact cells [34, 35].
With the finding of the first "endocannabinoid", the hypothesis of the existence of an "endogenous cannabinoid system", put forward for the first time after the molecular characterization and cloning of the "central" CB1 receptor in 1990 [36], acquired a further piece of evidence. The finding in 1993 of a "peripheral" cannabinoid receptor subtype, named CB2, in spleen cells, provided the molecular bases for the well known immunomodulatory actions of THC, thus seemingly completing the picture [37]. However, it was soon evident that ANA was much more effective at CB1 than CB2 receptors [37], where it rather behaved as a functional antagonist [38], thus suggesting that other metabolites might be present, in immunocompetent tissues and cells, whose function might be mediated by CB2 receptors.
DISCOVERY OF 2-AG AS AN ENDOCANNABINOID
A study carried out in 1995 [39] suggested for a congener of anandamide, palmitoylethanolamide (PEA), a possible function as agonist at CB2 receptors. PEA was found to inhibit serotonin release from antigen-challenged rat basophilic leukemia (RBL-2H3) cells and mastocytes at submicromolar doses, and to selectively displace high affinity cannabinoid ligands from RBL-2H3 cell membrane preparations. ANA did not exert this action but, on the contrary, antagonized the immunosuppressant effects of PEA and synthetic cannabinoids. Moreover, RBL-2H3 cells were shown to express CB2, but not CB1, cannabinoid receptors [39]. This evidence seemed to point to PEA as a possible agonist at CB2 receptors, and was supported by the previously reported anti-inflammatory effects (reviewed in [25]) of this compound, as well as by the finding that RBL-2H3 cells synthesize, uptake and degrade both PEA and ANA [39]. However, subsequent investigations, carried out using cells transfected with CB2 receptor complementary DNA, showed that PEA cannot compete with cannabinoid ligands for the binding to membrane preparations overexpressing CB2 receptors [40]. This suggested that the basophil/mast cell down-regulatory action of PEA was not mediated by any of the cannabinoid receptors described so far, and prompted the existence of other non-CB1-non-CB2 cannabinoid receptors, as well as of other ligands for the CB2 receptor.
While evidence for "CBn" cannabinoid receptors is still scattered and inconclusive, in 1995 Mechoulam and co-workers, by applying a binding assay-directed purification procedure to lipid extracts of canine gut, reported the isolation, structure elucidation and pharmacological characterization of a novel endogenous cannabimimetic substance [41]. The new putative "endocannabinoid" was an arachidonoyl ester rather than an amide, i.e., 2-AG. Apart from binding to both CB1- and CB2-containing cell membrane preparations, with Ki values, respectively, of 0.472 and 1.40 µM, 2-AG was found to be active in the "tetrad" of mice behavioral assays typical of psychoactive cannabinoids, i.e., a) the ring immobility test, where the time during which mice remain motionless on a ring is measured; b) the open field test, in which inhibition of locomotor activity is determined; c) hypothermia, and d) anti-nociception on a hot plate. Moreover, like ANA and THC, 2-AG inhibited forskolin-induced adenylate cyclase activation [41], and counteracted, although to a much lower extent compared to ANA and THC, the electrically-induced contractions of the isolated mouse vas deferens [41]. Indeed, the cannabimimetic properties of 2-AG could be predicted from its chemical structure, where two of the three functional groups thought to constitute the cannabinoid pharmacophore, i.e., the hydroxyl group and the n-pentyl alkyl chain [42], are present at almost the same distance as in ANA (Fig. 1). Moreover, a possible functional analogy between ANA and alkyl glycerol esters had been also suggested by the finding that the former metabolite is a partial agonist at the DAG binding site of PKC [43], and that acylethanolamide-phosphates and alkyl-lysophosphatidic acid have similar agonist activity towards the lysophosphatidic acid receptor [44]. Therefore, it was not surprising that 2-AG could exhibit cannabinoid-like properties, and this possibility had been indeed explored already in 1994, although, probably due to its degradation by lipases present in cell membrane preparations, only a weak cannabinoid receptor binding activity was originally observed for the monoglyceride [45].
Immediately after the isolation of 2-AG from canine gut, and in agreement with its behavioral properties [41], monoarachidonoyl-glycerol, as a mixture of the two 1- and 2-regio-isomers, and in amounts 500-1000 times higher than those of ANA, was found as the major component of rat brain monoacylglycerol fraction [46]. Since AA is mostly esterified in the sn-2 position of glycerol esters, the presence of the 1-isomer was suggested to be due to acyl migration from the sn-2 to the sn-1 position of the 2-isomer.
2-AG was also shown to bind, with a Ki = 2.4 µM, to rat brain synaptosomal preparations when these were treated with the esterase inhibitor diisopropyl fluorophosphate [46]. Later, a neuromodulatory role for the monoglyceride was strongly supported also by the finding of molecular mechanisms for its enzymatic degradation and Ca2+-dependent biosynthesis and release by mouse neuroblastoma N18TG2 cells ([47, 28] and below). Moreover, 2-AG was found to significantly affect the permeability to Ca2+ of both differentiated and undifferentiated hybrid (neuroblastoma × glioma) NG108-15 cells [48, 49]. Nanomolar concentrations of the cannabimimetic metabolite induced a rapid as well as transient elevation of intracellular Ca2+ in undifferentiated cells which was mimicked by the alkyl indole synthetic cannabinoid WIN 55212-2 but not by ANA, and was blocked by the selective CB1 antagonist SR 141716A. Furthermore, cross-desensitization to this effect was observed between 2-AG and WIN 55212-2, thus demonstrating that 2-AG-induced elevation of intracellular [Ca2+] was mediated by the cannabinoid receptor. Since the effect was only in part sensitive to the presence of EGTA, it was suggested that 2-AG might be acting by activating the release of Ca2+ from intracellular stores [48]. Conversely, 2-AG, in the low micromolar range of concentrations, was found to strongly inhibit the depolarization-induced influx of Ca2+ in differentiated NG108-15 cells [49], thus mimicking a response typical of ANA and cannabinoids. However, it was not possible to determine whether this inhibitory action of 2-AG was effected through cannabinoid receptors and N-type Ca2+ channels.
Interestingly, while 2-AG activation of Ca2+ influx in undifferentiated cells was not observed to a significant extent with other 2-acyl-glycerol species such as 2-palmitoyl-, 2-oleoyl-, 2-linoleoyl-, and 2-docosahexaenoyl-glycerol (which are inactive in cannabinoid receptor binding assays as well as in the mouse "tetrad" of behavioral tests [41]), the latter two compounds were almost equipotent with 2-AG in inhibiting high K+-induced Ca2+ influx in differentiated cells. A possible explanation for this apparent discrepancy is that polyunsaturated monoacylglycerols, at micromolar concentrations, can raise the levels of endogenous 2-AG by competing for the same inactivating lipase(s) (see next section). Finally, 2-AG has been recently described to inhibit long term potentiation in rat hippocampus [50], thus possibly sharing with ANA an impairing effect on memory [51].
Although its finding and pharmacological actions in the CNS suggests for 2-AG a role as an additional cannabimimetic neuromodulator, its possible regulatory function on peripheral cells selectively containing CB2 receptors, which bind only weakly to ANA [37, 38], may turn out to be even a more important one. Indeed, 2-AG, but not ANA, was found to inhibit the proliferation of B6C3F1 mouse splenocytes, and since of the two compounds only 2-AG was capable of inhibiting also the forskolin-induced activation of adenylate cyclase in the same cells, it is possible that the latter express CB2 receptors only [52]. Indeed, a recent study has shown that very little, if any, RNA encoding for the CB1 receptor is present in mouse spleen and several mouse and human splenic cells and T-cell lines, where CB2-like transcripts always seem to be predominant [53]. A primary role for 2-AG as immunomodulatory "endocannabinoid", at least in those immunocompetent cells which do not express CB1 receptors, is supported also by its recent isolation, together with several non-cannabimimetic monoacylglycerols, from canine spleen, which does not contain measurable amounts of ANA [54].
BIOSYNTHESIS AND INACTIVATION OF 2-AG
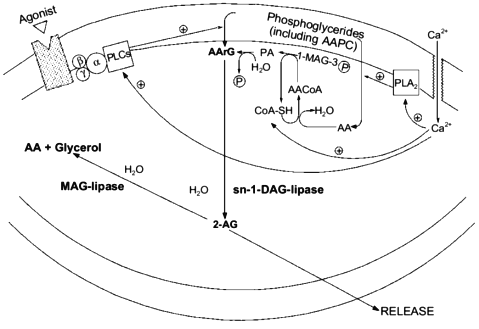
In order for a substance to be proposed as a mediator in a certain organism, molecular mechanisms must be found to be available to that organism allowing the biosynthesis and inactivation of the putative mediator. Although some papers had previously described the stimulus-induced formation of monoarachidonoyl-glycerol like-metabolites from either peripheral cells (for example, see [3]) or dorsal root ganglia [5], the report that ionomycin stimulation can trigger 2-AG biosynthesis and release from mouse neuroblastoma N18TG2 cells in a fashion depending on extracellular Ca2+ [47] was the first of this kind to rigorously demonstrate the chemical nature of the glyceride produced as that of the 2-isomer of monoarachidonoyl-glycerol. Moreover, by treating the cells with several exogenous phospholipases, it was possible to observe the release of 2-AG only concomitantly with the formation of sn-2-AA-containing DAGs (AArGs). Accordingly, neuroblastoma cell homogenates were found to contain an sn-1-DAG-lipase activity capable of efficiently converting AArG into 2-AG. The major source for the stimulus-induced mobilization of AArG in cells is the PLC-catalyzed hydrolysis of sn-2-AA-containing PC and PI. However, ionomycin-induced 2-AG formation was not inhibited by the PLC inhibitor neomycin sulfate, and was potentiated by pre-treatment of cells with exogenous PLA2, which depletes sn-2-AA from membrane phosphoglycerides, thus decreasing considerably the amounts of phospholipid precursors for AArG [47]. These data seem to suggest that ionomycin, which directly activates Ca2+ influx into cells and by-passes PLC-mediated Ca2+ mobilization, induce 2-AG formation by leading to de novo AArG synthesis (from AA-CoA and glycerol-3-phosphate and via phosphatidic acid (PA), see Fig. 2) and not to AArG release from membrane phosphoglycerides. This may also explain why an augmented availability of free arachidonate, that alone was not sufficient to cause 2-AG biosynthesis, remarkably facilitated the ionomycin-induced effect [47]. However, mouse neuroblastoma cell homogenates were also found to contain [28] two distinct (being, respectively, insensitive and sensitive to EGTA) PLC-like enzymes capable of slowly but significantly converting sn-2-AA-containing sn-1-lyso-PC or PC, respectively, into 2-AG or AArG plus 2-AG (Fig. 2). Accordingly, a recent study carried out in cultured rat cortical neurons and electrically-stimulated hippocampal slices, confirmed that 2-AG synthesis may occur through the sequential action of phospholipase C and sn-1-DAG-lipase [50]. Therefore, central neurons may produce 2-AG also upon stimulation of G-protein coupled PLC enzymes, as previously observed in Swiss 3T3 fibroblasts stimulated with platelet derived growth factor [3] and dorsal root ganglia treated with bradykinin [5].
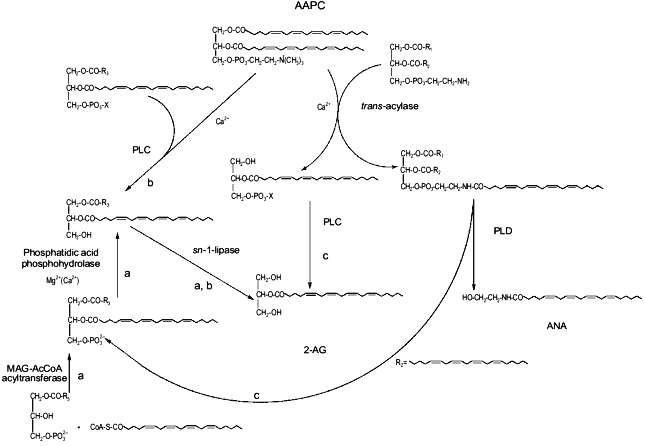
This possibility may create intriguing interactions between the biosynthesis of 2-AG and ANA. In fact, one of the possible ultimate precursors of the latter metabolite is a particular and not very abundant PC species, AAPC, which serves as arachidonate donor for the trans-acylase-mediated formation of ANA direct precursor, N-ArPE (Fig. 3 and [26-29]). The by-product of this Ca2+-dependent trans-acylation is sn-1-lyso-2-arachidonoyl-PC, which can be converted into 2-AG by the EGTA-insensitive PLC-like enzyme described above [28]. It is unlikely that this enzyme is the same as the 1-lyso-PI-specific PLC previously characterized in rat brain synaptosomes [14], which was reported to recognize to a very low extent also lysoPC species, but was also shown to be sensitive to the presence of EDTA in the incubation medium [55]. On the other hand, instead of contributing to N-ArPE and ANA formation, AAPC could be converted into the corresponding AArG (and, subsequently, into 2-AG) through the catalytic action of the EGTA-sensitive PLC-like enzyme found in mouse neuroblastoma cells [28]. Therefore, along with a Ca2+-dependent and PLC-independent pathway, enzymatic mechanisms also exists for the Ca2+-dependent and PLC-dependent formation of 2-AG from sn-2-AA-containing (lyso)phospholipids. While in the former case 2-AG biosynthesis would occur independently from ANA formation, and might depend also, but not only, on the availability of AA-CoA (Fig. 2), the latter pathways may lead to the monoglyceride either concomitantly or in competition with ANA (Fig. 3).Fig. 2. Possible pathways for 2-arachidonoyl-glycerol biosynthesis and metabolism in mouse neuroblastoma cells (based on references [5, 28, 47]). 2-AG, 2-arachidonoyl-glycerol; PLA2, phospholipase A2; AA, arachidonic acid; 1-MAG-3P, sn-1-acylglycerol-3-phosphate; AACoA, arachidonoyl-coenzyme A; CoA-SH, reduced coenzyme A; PA, phosphatidic acid; AArG, sn-1-acyl-2-arachidonoyl-glycerol; DAG, diacylglycerol; MAG, monoacylglycerol; PLC, phospholipase C; AAPC, sn-1,2-diarachidonoyl-phosphatidylcholine.
Finally, AArG precursors for 2-AG may be formed from the enzymatic hydrolysis of sn-2-AA-containing phosphatidic acid (PA) coming also from the PLD-mediated conversion of N-ArPE into ANA. Again, in this case the two "endocannabinoids" may be produced simultaneously (Fig. 3).Fig. 3. Possible biosynthetic connections between anandamide and 2-arachidonoyl-glycerol in mouse neuroblastoma cells (based on references [28, 47]). AAPC, sn-1,2-diarachidonoyl-phosphatidylcholine; MAG-AcCoA, monoacylglycerol-3-phosphate:acyl-coenzyme A. a) Phospholipase C-independent pathway occurring independently from anandamide biosynthesis; b) phospholipase C-dependent pathway occurring partly in competition with anandamide biosynthesis; c) phospholipase C-dependent pathway occurring concomitantly with anandamide biosynthesis.
Therefore, together with the previously described [14] sequential action of PI-selective phospholipase A1 and lysoPI-specific PLC (which, however, is not likely to underlie the Ca2+-dependent release of 2-AG since the former enzyme was found to be insensitive to variations of [Ca2+]), several routes may contribute to 2-AG biosynthesis in neurons. This may explain why, in rat brain, the levels of the monoglyceride are much higher than those of ANA. Due to the possible intersections of these routes with the pathway leading to ANA biosynthesis, it will be interesting to investigate in the future whether the same stimuli, such those leading to phospholipase D and/or adenylate cyclase activation [29], which facilitate N-ArPE and ANA formation in neurons, also affect 2-AG levels.
While the degradation of ANA has been thoroughly investigated, leading to the molecular characterization, cloning and expression [56] of "anandamide amidohydrolase", subsequently renamed "fatty acid amide hydrolase" (FAAH), the enzyme responsible for the hydrolysis of the amide bond of ANA as well as other bioactive long chain fatty acid amides [56-59, 39], very little is known about the enzymatic activity catalyzing the hydrolysis of 2-AG to glycerol and AA. This activity was found in mitochondrial (10,000g pellet), microsomal, and cytosolic fractions of mouse neuroblastoma N18TG2 cells [47], which seemed to suggest the presence of several esterases all capable of recognizing 2-AG as a substrate. Therefore, differently from ANA, there is no evidence to date for the existence of a specific enzyme deputed to the inactivation of 2-AG.
Evidence for at least a certain specificity for the enzymatic hydrolysis of 2-AG came from a more recent study ([54] and V. Di Marzo and T. Bisogno, unpublished data) where: a) 10,000g pellet fractions prepared from RBL-2H3 and N18TG2 cells were found to hydrolyze both 1- and 2-AG, and b) monoacylglycerols with fatty acid chains with at least two 1,4-diene double bonds, such as 1- and 2-linoleoyl- and 1-linolenoyl-glycerol, but not the palmitoyl- and myristoyl-analogs, were shown to inhibit monoarachidonoyl-glycerol hydrolysis. This suggests that 2-AG hydrolyzing enzymatic activity(ies) is (are) at least selective towards polyunsaturated monoacylglycerols, even though it (they) can recognize as substrates both 1- and 2-regio-isomers of 2-AG. These activity(ies) may be due, in part, to the same enzyme(s) previously reported to catalyze AA liberation from 2-AG [10-12], but not to the nonspecific ester hydrolase, which recognizes also saturated monoglycerides and cholesterol esters, nor to the pancreatic MAG-lipase, which is unable to hydrolyze the ester bond at position sn-2 [1].
The study carried out in N18TG2 cells confirmed, however, the presence of nonspecific esterases, since the hydrolysis of [3H]2-AG was only partly (less than 40%) inhibited by an excess of unlabelled 2-AG, whereas the degree of selectivity of the esterase(s) from the "peripheral" RBL-2H3 cell line appeared to be remarkably higher (74% inhibition of 4 µM [3H]2-AG hydrolysis by 100 µM 2-AG). Moreover, it was shown that intact RBL-2H3 cells are capable of time-dependently sequestering exogenous [3H]2-AG from the incubation medium. The clearance of 2-AG was half-maximal after 6 min, and this was the result of uptake and degradation by cells, as shown by the finding of increasing amounts of cell-associated [3H]2-AG and of [3H]AA in the medium. Interestingly, 2-linoleoyl-glycerol, but not 2-palmitoyl-glycerol, was found to inhibit also 2-AG sequestration by intact cells, and, therefore, to increase considerably the amounts of free 2-AG available for cannabinoid receptor activation, by preventing both its uptake and hydrolysis [54]. This finding suggested that non-cannabimimetic monoacylglycerols, that are found to accompany 2-AG in several peripheral tissues [41, 54], may facilitate the action of 2-AG by prolonging its half-life. Furthermore, 2-AG homologs, and in particular monopalmitoyl-glycerol, that does not inhibit 2-AG inactivation, caused a 4-8-fold potentiation of 2-AG binding to CB2 receptors [54], thus rendering the affinity of this metabolite for the "peripheral" receptor higher than that reported for ANA [37]. This led to propose that long chain monoacylglycerols may operate an "entourage" effect on 2-AG and enhance its pharmacological actions by both inhibiting 2-AG degradation and increasing the efficiency of 2-AG interaction with cannabinoid receptors. This hypothesis was confirmed when mixtures of 2-acyl-glycerols were tested in the "tetrad" of behavioral tests in mice. In fact, when 2-AG at a concentration not sufficient to elicit full cannabimimetic responses (1 mg/kg body weight) was co-administered with a mixture of 2-palmitoyl- and 2-linoleoyl-glycerol (which alone have no effect in these behavioral tests) in a molar ratio to 2-AG similar to that usually found in tissues (i.e., 5:10:1), analgesia, hypothermia, and inhibition of motor behavior were observed [54]. This type of "entourage" effect has been reported [60] also for ANA by fatty acid amides, such as oleamide, oleoylethanolamide, and linoleoylethanolamide (which inhibit ANA hydrolysis by competing with FAAH [57, 61]), and may represent a physiologically relevant mechanism for the regulation of "endocannabinoid" action by naturally occurring, non-cannabimimetic "endocannabinoid" analogs.
Finally, the conversion of 2-AG into the 1-isomer may be regarded as a form of metabolism. AA is mostly esterified to the sn-2 position of phosphoglycerides (although a little amount can be found also on the sn-1 position, as in AAPC) and DAGs, and, therefore, the finding of 1-AG is very likely to be due to acyl migration from the sterically hindered sn-2 position to the more stable sn-1 or sn-3 positions, a non-enzymatic process typical of di- and monoglycerides. It is difficult to assess how much of the formation of 1-AG is due to an artifact of the extraction and purification procedure, and how much occurs in living cells following the formation of 2-AG. In any case, since 2-AG does not differ significantly from its 1-isomer in cannabinoid receptor binding activity [50], this process would probably not represent a route for the inactivation of the "endocannabinoid".
Probably due also to the relatively recent discovery of the cannabimimetic properties of 2-AG, still very little is known on their possible physiological importance. From the knowledge currently available it is possible to hypothesize for this compound a role as an auxiliary "endocannabinoid" at CB1 receptors and of primary "endocannabinoid" at CB2 receptors. In the first case, 2-AG, synthesized following neuronal stimulation either independently or concomitantly to ANA, might reinforce the action of the latter, which has been suggested to act as a partial cannabinoid agonist in the CNS [62]. Evidence accumulated so far indicates that such reinforcing action may take place during the control of motor function, pain perception, body temperature, and memory [41, 50, 54]. In the second case, the monoacylglycerol, especially if synthesized together with a series of congeners, might efficiently effect CB2-mediated immunomodulatory functions on lymphocytes, where the action of ANA seems to be restricted to a few CB1-containing cell types. A CB2-mediated functional action for 2-AG can be foreseen also in the CNS due to the recent finding of CB2 receptors coupled to inhibition of cAMP formation in rat microglia [63].
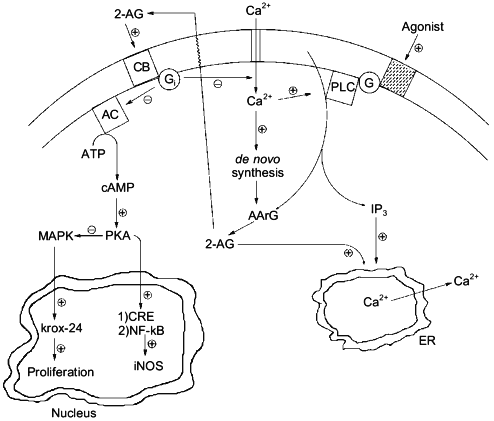
Experiments carried out in the past, where agonist-induced formation of 2-AG was recorded but often over-looked, should be re-evaluated on the basis of the pharmacological activities recently described for 2-AG and/or CB receptor activation both in neurons and immune cells, e.g., inhibition of adenylate cyclase and protein kinase A (both CB1 and CB2), modulation of intracellular [Ca2+] (CB1), activation of mitogen-activated protein kinase (both CB1 and CB2) and up-regulation of the growth-related gene krox-24 (CB2), inhibition of nuclear factor .B and of cAMP-response binding element activity with subsequent down-regulation of inducible NO synthase (CB2), etc. (Fig. 4 and [64, 65]). Structure--activity relationship studies, using cannabinoid receptor binding tests for probing the activity of structural analogs, have not been yet performed on 2-AG. However, important pieces of information on the structural requirements for pharmacological activity of the monoglyceride can be obtained by the several structure--activity relationship studies so far reported for ANA (for example [50]), where arachidonoyl esters have sometimes been studied. It will be important, for the design of stable and more potent 2-AG analogs, to take into account the previous observation that arachidonoyl-ethylene-glycol has the same potency as 2-AG in binding assays conducted with rat brain synaptosomal membranes [46]. Also, it will be soon possible to assess which of the peripheral actions of 2-AG are due to CB2-receptor activation by using the recently developed CB2 antagonist SR144528 [66].
In conclusion, further research will still be needed on 2-AG in order to definitively bring this metabolite to the limelight of the bioactive lipid research forum. Selective inhibitors of its degradation will have to be developed in order to gain additional data on its pharmacological activity both in vivo and in vitro, and renewed efforts will have to be made towards the clarification of which of the several biosynthetic pathways so far proposed are actually responsible for its physiological formation and under which regulative mechanisms. Due to the widespread occurrence of 2-AG in cells, these studies are likely to lead to surprising insights in both animal and plant physiology.Fig. 4. Possible intracellular, cannabinoid receptor-mediated actions of 2-arachidonoyl-glycerol (2-AG): interactions with inositol-tris-phosphates (IP3) and cyclic AMP (cAMP). By counteracting the depolarization-induced influx of Ca2+ into neurons [49], 2-AG would operate a negative feed-back control on its own synthesis [47]. By inducing, through activation of CB1 by receptors, the release of Ca2+ from the endoplasmic reticulum (ER) [48], 2-AG might potentiate the action of inositol-phosphates, which may be produced together with the monoglyceride [3, 5]. Finally, by activating CB2 receptors and subsequently inhibiting cAMP formation, 2-AG might on the one hand release the inhibition of mitogen-activated protein kinase (MAPK), thus leading to the activation of krox-24 [64], and, on the other hand, counteract the protein kinase A (PKA)-mediated activation of cAMP-response binding element (CRE), nuclear factor .B (NF-.B), and, ultimately, inducible nitric oxide synthase (iNOS) [65]. PLC, phospholipase C; AArG, sn-1-acyl-2-arachidonoyl-glycerol; G and Gi, G-proteins; CB, either CB1 or CB2 cannabinoid receptor; AC, adenylate cyclase.
The author wishes to thank Prof. R. Mechoulam (The Hebrew University, Jerusalem, Israel) and Dr. T. Sugiura (Teikyo University, Sagamiko, Kanagawa, Japan) for sharing their unpublished data and Drs. L. De Petrocellis, T. Bisogno, D. Melck, and N. Sepe as co-authors of several papers on 2-AG. Financial aid from the Human Frontiers in Science Program Organization (RG 26/95) is also acknowledged.
REFERENCES
1.Vance, D. E., and Vance, J. (1991) Biochemistry
of Lipids, Lipoproteins and Membranes, Elsevier, Amsterdam.
2.Gerwich, W. H., Moghaddam, M. F., and Hamberg, M.
(1991) Arch. Biochem. Biophys., 290, 436-444.
3.Hasegawa-Sasaki, H. (1985) Biochem. J.,
232, 99-109.
4.Prescott, S. M., and Majerus, P. W. (1983) J.
Biol. Chem., 258, 764-769.
5.Allen, A. C., Gammon, C. M., Ousley, A. H.,
McCarthy, K. D., and Morell, P. (1992) J. Neurochem.,
58, 1130-1139.
6.Gobbetti, A., and Zerani, M. (1993) Cell
Signalling, 5, 63-67.
7.Rapuano, B., and Bockman, R. S. (1991) Biochim.
Biophys. Acta, 1091, 374-384.
8.Bell, R. L., Kennerly, D. A., Stanford, N., and
Majerus, P. W. (1979) Proc. Natl. Acad. Sci. USA, 76,
3238-3241.
9.Di Marzo, V. (1995) Prostaglandins Leukot.
Essent. Fatty Acids, 53, 239-254.
10.Farooqui, A. A., Anderson, D. K., Flynn, C.,
Bradel, A., Means, E. D., and Horrocks, L. A. (1990) Biochem.
Biophys. Res. Commun., 166, 1001-1009.
11.Dixon, J. F., and Hokin, L. E. (1984) J. Biol.
Chem., 259, 14418-14425.
12.Gammon, C. M., Allen, A. C., and Morell, P.
(1989) J. Neurochem., 53, 95-101.
13.Askari, A., Xie, Z., Wang, Y., Periyasamy, S.,
and Huang, W. (1991) Biochim. Biophys. Acta, 1069,
127-130.
14.Ueda, H., Kobayashi, T., Kishimoto, M., Tsutsumi,
T., and Okuyama, H. (1993) J. Neurochem., 61,
1874-1881.
15.Devane, W. A., Hanus, L., Breuer, A., Pertwee, R.
G., Stevenson, L. A., Griffin, G., Gibson, D., Mandelbaum, A., Etinger,
A., and Mechoulam, R. (1992) Science, 258,
1946-1949.
16.Fride, E., and Mechoulam, R. (1993) Eur. J.
Pharmacol., 231, 313-314.
17.Crawley, J. N., Corwin, R. L., Robinson, J. K.,
Felder, C. C., Devane, W. A., and Axelrod, J. (1993) Pharmacol.
Biochem. Behav., 46, 967-972.
18.Smith, P. B., Compton, D. R., Welch, S. P.,
Razdan, R. K., Mechoulam, R., and Martin, B. R. (1994) J. Pharmacol.
Exp. Ther., 270, 219-227.
19.Felder, C. C., Briley, E. M., Axelrod, J.,
Simpson, J. T., Mackie, K., and Devane, W. A. (1993) Proc. Natl.
Acad. Sci. USA, 90, 7656-7660.
20.Vogel, Z., Barg, J., Levy, R., Saya, D., Heldman,
E., and Mechoulam, R. (1993) J. Neurochem., 61,
352-355.
21.Di Marzo, V., De Petrocellis, L., Bisogno, T.,
and Maurelli, S. (1995) J. Drug Dev. Clin. Pract., 7,
199-219.
22.Shohami, E., Weidenfeld, J., Ovadia, H., Vogel,
Z., Hanus, L., Fride, E., Breuer, A., Ben-Shabat, S., Sheskin, T., and
Mechoulam, R. (1997) CNS Drug Rev., 4, 429-451.
23.Dewey, W. L. (1986) Pharmacol. Rev.,
38, 151-178.
24.Di Marzo, V., Fontana, A., Cadas, H., Schinelli,
S., Cimino, G., Schwartz, J. C., and Piomelli, D. (1994) Nature,
372,686-691.
25.Schmid, H. H. O., Schmid, P. C., and Natarajan,
V. (1990) Prog. Lipid Res., 29, 1-43.
26.Sugiura, T., Kondo, S., Sukagawa, A., Tonegawa,
T., Nakane, S., Yamashita, A., Ishima, Y., and Waku, K. (1996) Eur.
J. Biochem., 240, 53-62.
27.Cadas, H., Gaillet, S., Beltramo, M., Venance,
L., and Piomelli, D. (1996) J. Neurosci.,
16,3934-3942.
28.Di Marzo, V., De Petrocellis, L., Sugiura, T.,
and Waku, K. (1996) Biochem. Biophys. Res. Commun., 227,
281-288.
29.Cadas, H., di Tomaso, E., and Piomelli, D. (1997)
J. Neurosci., 17, 1226-1242.
30.Sugiura, T., Kondo, S., Sukagawa, A., Tonegawa,
T., Nakane, S., Yamashita, A., and Waku, K. (1996) Biochem. Biophys.
Res. Commun., 218, 113-117.
31.Bisogno, T., Maurelli, S., Melck, D., De
Petrocellis, L., and Di Marzo, V.(1997) J. Biol. Chem.,
272, 3315-3323.
32.Di Marzo, V., De Petrocellis, L., Sepe, N., and
Buono, A.(1996) Biochem. J., 316, 977-984.
33.Bisogno, T., Ventriglia, M., Mosca, M., Milone,
A., Cimino, G., and Di Marzo, V. (1997) Biochim. Biophys. Acta,
1345, 338-348.
34.Kruszka, K. K., and Gross, R. W. (1994) J.
Biol. Chem., 269, 14345-14348.
35.Devane, W. A., and Axelrod, J. (1994) Proc.
Natl. Acad. Sci. USA, 91, 6698-6701.
36.Matsuda, L. A., Lolait, S. J., Brownstein, M. J.,
Young, A. C., and Bonner, T. I. (1990) Nature, 346,
561-564.
37.Munro, S., Thomas, K. L., and Abu-Shaar, M.
(1993) Nature, 365, 61-65.
38.Bayewitch, M., Avidor-Reiss, T., Levy, R., Barg,
J., Mechoulam, R., and Vogel, Z.(1995) FEBS Lett., 375,
143-147.
39.Facci, L., Dal Toso, R., Romanello, S., Buriani,
A., Skaper, S. D., and Leon, A.(1995) Proc. Natl. Acad. Sci.
USA, 92, 3376-3380.
40.Sheskin, T., Hanus, L., Slager, J., Vogel, Z.,
and Mechoulam, R. (1997) J. Med. Chem., 40, 659-667.
41.Mechoulam, R., Ben-Shabat, S., Hanus, L.,
Ligumsky, M., Kaminski, N. E., Schatz, A. R., Gopher, A., Almog, S.,
Martin, B. R., Compton, D. R., Pertwee, R. G., Griffin, G., Bayewitch,
M., Barg, J., and Vogel, Z. (1995) Biochem. Pharmacol.,
50, 83-90.
42.Martin, B. R., Thomas, B. F., and Razdan, R. K.
(1995) in Cannabinoid Receptors (Pertwee, R. G., ed.) Academic
Press, London, pp. 35-79.
43.De Petrocellis, L., Orlando, P., and Di Marzo, V.
(1995) Biochem. Mol. Biol. Int., 36, 1127-1133.
44.Sugiura, T., Tokumura, A., Gregory, L., Nouchi,
T., Weintraub, S. T., and Hanahan, D. J. (1994) Arch. Biochem.
Biophys., 311, 358-368.
45.Sugiura, T., Itoh, K., Waku, K., and Hanahan, D.
J. (1994) in Proc. Japan. Conf. on the Biochemistry of Lipids
[in Japanese], 36, 71-74.
46.Sugiura, T., Kondo, S., Sukagawa, A., Nakane, S.,
Shinoda, A., Itoh, K., Yamashita, A., and Waku, K. (1995) Biochem.
Biophys. Res. Commun., 215, 89-97.
47.Bisogno, T., Sepe, N., Melck, D., Maurelli, S.,
De Petrocellis, L., and Di Marzo, V.(1997) Biochem. J.,
322, 671-677.
48.Sugiura, T., Kodaka, T., Kondo, S., Tonegawa, T.,
Nakane, S., Kishimoto, S., Yamashita, A., and Waku, K. (1996)
Biochem. Biophys. Res. Commun., 229, 58-64.
49.Sugiura, T., Kodaka, T., Kondo, S., Tonegawa, T.,
Nakane, S., Kishimoto, S., Yamashita, A., and Waku, K. (1997)
Biochem. Biophys. Res. Commun., 233, 207-210.
50.Stella, N., Schweitzer, P., and Piomelli, D.
(1997) Nature, 388, 773-778.
51.Mallet, P. E., and Beninger, R. J. (1996)
Behav. Pharmacol., 7, 276-284.
52.Lee, M., Yang, K. H., and Kaminski, N. E. (1995)
J. Pharmacol. Exp. Ther., 275, 529-536.
53.Schatz, A. R., Lee, M., Condie, R. B., Pulaski,
J. T., and Kaminski, N. E. (1997) Toxicol. Appl. Pharmacol.,
142, 278-287.
54.Ben-Shabat, S., Fride, E., Sheskin, T., Tamiri,
T., Rhee, M.-H., Vogel, Z., Bisogno, T., De Petrocellis, L., Di Marzo,
V., and Mechoulam, R., Mol. Pharmacol., submitted.
55.Tsutsumi, T., Kobayashi, T., Ueda, H., Yamauchi,
E., Watanabe, S., and Okuyama, H. (1994) Neurochem. Res.,
19, 399-406.
56.Cravatt, B. F., Giang, D. K., Mayfield, S. P.,
Boger, D. L., Lerner, R. A., and Gilula, N. B. (1996) Nature,
384, 83-87.
57.Maurelli, S., Bisogno, T., De Petrocellis, L., Di
Luccia, A., Marino, G., and Di Marzo, V.(1995) FEBS Lett.,
377, 82-86.
58.Ueda, N., Kurahashi, Y., Yamamoto, K., and
Yamamoto, S. (1997) Adv. Exp. Med. Biol., 407,
323-328.
59.Koutek, B., Prestwich, G. D., Howlett, A. C.,
Chin, S. A., Salehani, D., Akhavan, N., and Deutsch, D. G.(1994) J.
Biol. Chem., 269, 22937-22940.
60.Fride, E., Bisogno, T., Di Marzo, V., Bayewitch,
M., Vogel, Z., and Mechoulam, R. (1997) in Abstract Book of the
Society for Neuroscience Annual Meeting, New Orleans, 25-30
October.
61.Di Tomaso, E., Beltramo, M., and Piomelli, D.
(1996) Nature, 382, 677-678.
62.Mackie, K., Devane, W. A., and Hille, B. (1993)
Mol. Pharmacol., 44, 498-503.
63.Kearn, C. S., and Hillard, C. J. (1997) in
Abstract Book of the 7th ICRS Conference, Stone Mountain,
Atlanta, Georgia, 19-22 June, p. 61.
64.Jeon, Y. J., Yang, K. H., Pulaski, J. T., and
Kaminski, N. E. (1996) Mol. Pharmacol., 50, 334-341.
65.Bouaboula, M., Poinot-chazel, C., Marchand, J.,
Canat, X., Bourrie, B., Rinaldi-Carmona, M., Calandra, B., Le Fur, G.,
and Casellas, P. (1996) Eur. J. Biochem., 237,
704-711.
66.Barth, F., Rinaldi-Carmona, M., Millan, J.,
Derocq, J.-M., Bouaboula, M., Casellas, P., Congy, C., Oustric, D.,
Sarran, M., Calandra, B., Portier, M., Shire, D., Breliere, J.-C., and
Le Fur, G. (1997) in Abstract Book of the 7th ICRS Conference,
Stone Mountain, Atlanta, Georgia, 19-22 June, p. 11.