DNA- and RNA-Hydrolyzing Antibodies from the Blood of Patients with Various Forms of Viral Hepatitis
A. G. Baranovsky,1 V. G. Matushin,2 A. V. Vlassov,1 V. G. Zabara,3 V. A. Naumov,3 R. Giege,4 V. N. Buneva,1 and G. A. Nevinsky1,5
1Novosibirsk Institute of Bioorganic Chemistry, Siberian Branch of Russian Academy of Sciences, pr. Lavrent'eva 8, Novosibirsk, 630090 Russia; fax: (3832) 35-1665; E-mail: nev@modul.bioch.nsk.su2Novosibirsk State University, pr. Pirogova 2, Novosibirsk, 630090 Russia.
3Army Hospital No. 333, ul. Voinskaya 24, Novosibirsk, 630017 Russia.
4Institut de Biologie Moleculaire et Cellulaire du CNRS, 15, rue Rene Descartes, Strasbourg 67000, France.
5To whom correspondence should be addressed.
Submitted July 17, 1997.
Antibodies (Abs) hydrolyzing proteins, DNA, and RNA are detected in the blood of patients with various autoimmune diseases. In the present work, homogeneous preparations of IgG Abs from the blood of the healthy donors as well as patients with A, B, C, and delta types of viral hepatitis, influenza, pneumonia, tuberculosis, tonsillitis, duodenal ulcer, and some types of cancer were purified. For the first time, the fraction of IgG and its Fab fragments of patients with viral hepatitis were shown to have high DNA- and RNA-hydrolyzing activity. In case of Abs from the healthy donors and patients with other diseases, high activity of Abs was not detected. The data obtained by various methods indicate that the activity of hepatitis Abs is an intrinsic property of the immunoglobulins. The relative rates of hydrolysis of cCMP, poly(U), poly(A), poly(C), and tRNAPhe by hepatitis Abs were compared with those of RNAse A and other RNAses from human blood. Significant differences in activities of Abs and nucleases in hydrolysis of model substrates were demonstrated. Thus, catalytically active Abs can appear in the blood of patients not only with autoimmune disorders, but with viral diseases as well.
KEY WORDS: catalytically active antibodies, abzyme, human blood, hepatitis, ribonuclease, RNA hydrolysis, DNA hydrolysis.
Abbreviations: Ab) antibody; ELE) exanthematous lupus erythematosus; AIT) autoimmune thyroiditis or Hashimoto's disease; PA) polyarthritis; NA) nucleic acid.
Most catalytically active Abs6 or so-called abzymes are Abs
raised against a stable analog of the transition state complex of a
catalytic reaction that is used as hapten [1-6]. At present, over 50 reactions have been described
to be catalyzed by Abs.
Natural abzymes were first demonstrated in the human body in the blood of patients with bronchial asthma [7]. These auto-Abs specifically cleave an oligopeptide neurohormone. Later DNA-hydrolyzing Abs were detected in the blood of patients with ELE. Protein-hydrolyzing Abs have been described in AIT [8] and DNA- and RNA-hydrolyzing Abs have been detected in certain autoimmune disorders including ELE [9-14], PA, and AIT [15]. The breast milk of the apparently healthy women contains Abs with protein kinase and RNA- and DNA-hydrolyzing activities [16-20].
ELE patients usually have increased blood level of DNA and anti-DNA Abs. This phenomenon was the starting point of investigation of DNA-hydrolyzing Abs in ELE patients [9-12]. Increased levels of DNA and anti-DNA Abs have been observed in other autoimmune disorders including PA and AIT [21]; our previous data are in good agreement with this since DNA- and RNA-hydrolyzing Abs are present in the blood of these patients [15]. The immune status is altered in patients infected with immunodeficiency virus. From the immune system point of view, such infection is similar to autoimmune disorders because DNA and anti-DNA Abs appear in the blood [22]. Hence, it is not surprising that immunodeficiency syndrome in humans induces accumulation of DNA-hydrolyzing abzymes [23].
It is not known whether infection with hepatitis virus develops similarly to autoimmune process. However, this infection is associated with autoimmune humoral and cellular reactions; in particular, tissue-specific and non-specific autoantigens appear in the blood of hepatitis patients (similar to autoimmune disorders) [24]. Considering this, the levels of IgG-abzymes which cleave NA was evaluated in the blood of 49 patients with various forms of viral hepatitis. The blood of apparently healthy donors and patients with other disorders (influenza, pneumonia, tuberculosis, tonsillitis, duodenal ulcer, and some types of cancer) was taken for comparison. It is demonstrated that only Abs from the blood of patients with various forms of viral hepatitis have high NA-hydrolyzing activity.
MATERIALS AND METHODS
Isolation of Antibodies. Blood of healthy donors and patients was incubated for several hours to achieve complete coagulation. Blood serum was carefully separated from the clot and centrifuged. An equal volume of 50 mM Tris-HCl, pH 7.5, containing 100 mM NaCl (buffer A) was added to the supernatant. Proteins were precipitated with ammonium sulfate (50% saturation), incubated for 1 h at 0°C, and centrifuged; the pellet was redissolved in buffer A corresponding to the volume of the initial serum and loaded onto a protein A-Sepharose column (6.5 × 30 mm) equilibrated with buffer A.
Proteins that did not interact with the matrix were washed with buffer A to zero absorption. To destroy the non-covalent complexes of protein A with non-specifically bound proteins, the column was sequentially washed with buffer A containing 1% Triton X-100 and 0.3 M NaCl (5 ml), buffer A to zero absorption (5 ml), and 0.1 M sodium citrate (pH 5.0). Abs were eluted with 0.1 M glycine buffer (pH 2.6) and immediately neutralized with 1 M Tris-HCl (pH 8). The Abs were dialyzed against 500 volumes of buffer A for 10 h.
Purified Abs were gel-filtered through a Toyopearl HW-55 column (10 × 290 mm) as described [13, 15, 18]. The column was equilibrated with buffer A and 0.1 ml of Ab solution was loaded and eluted with the same buffer (0.2 ml/min). Abs were gel-filtered in acidic buffer (pH 2.6) in a similar manner. Abs were incubated for 1 h in the same buffer at 30°C prior to gel filtration as described previously [13, 15, 18].
Chromatography of Abs on DNA-cellulose was the final purification stage [13, 15].
SDS-PAGE Analysis of Abs. Homogeneity of the IgG preparation was assayed at each purification stage by SDS-PAGE under non-dissociating and dissociating conditions by the method of Laemmli [13, 15, 18, 25]. In the absence of 2-mercaptoethanol, electrophoresis was performed using 7-12% gradient gel and in its presence 12% gel was used. Proteins were stained with AgNO3 following electrophoresis [26]. Additional identification of type of Abs and their subunits was performed by immunoanalysis using various mono- and polyclonal Abs as described [27].
Preparation of IgG Fragments. To prepare the Fab fragments, IgG were hydrolyzed with papain as described [28, 29]. Fab fragments were purified by sequential chromatography of the Abs on protein A-Sepharose (as described above for Ab purification) and CM-Trisacryl columns [15]. Homogeneity of Fab fragments of IgG was demonstrated by electrophoresis as described above.
DNA Hydrolysis. Activity of Abs was evaluated by measuring the extent of transformation of supercoiled DNA (plasmid pBR-322) into circular nicked and linear forms and into oligonucleotides with various lengths. Reaction medium (20 µl) contained 25 mM Tris-HCl buffer (pH 7.5), 50 mM NaCl, 5 mM MgCl2, 0.5 mM EDTA, 10-20 µg/ml plasmid pBR-322 DNA, and 0.05-0.3 mg/ml Abs. Samples were incubated for 2 h (or 12 h in certain experiments as indicated) at 37°C and 4 µl of bromophenol blue dye were added to the reaction medium; samples were electrophoresed through a 0.8% agarose gel. DNA was stained with ethidium bromide (0.5 µg/ml).
Hydrolysis of Homopolynucleotides and cCMP. The hydrolysis rate of cCMP and homoribopolynucleotides (20°C) was assayed by the increase in optical density (linear part of kinetic curve) at 292 and 280 nm, respectively (Specord M40, Germany), similar to RNAse A assay [30, 31]. The reaction medium (0.2 ml; 1 mm thick cell) contained 50 mM Tris-HCl buffer (pH 7.5), 1 mg/ml polynucleotide or 1 mM cCMP. The reaction was initiated by addition of Abs or 1.0 µM RNAse A.
Preparation of 3´-Labeled tRNAPhe. tRNA was prepared and its 3´-end labeled as described [32, 33]; the procedure involved addition of CCA-terminal sequence using CTP, [alpha-32P]ATP, and (ATP,CTP):tRNA nucleotidyl transferase after partial hydrolysis of 3´-end of tRNA by snake venom phosphodiesterase. Two volumes of dye-containing 8 M urea were added to the labeled tRNA solution and samples were electrophoresed through 8% polyacrylamide gel. After separation, the labeled tRNA-containing fragment of the gel was excised and eluted by incubation for 10 h at 4°C in 300 µl of 0.5 M sodium acetate buffer (pH 5.5) containing 0.1% SDS, 1 mM EDTA, 50 µl phenol, and carrier tRNA (100 µg). The aqueous phase containing tRNA was removed after incubation, supplemented with 3 volumes of ethanol, and incubated for 4 h at -20°C, and the tRNA was pelleted by centrifugation. Pelleted tRNA was washed with 0.5 volumes of ethanol, dried, and dissolved in water.
Hydrolysis of tRNA by Ab Preparations. Hydrolysis of tRNA was performed in 20 µl reaction medium at 37°C for 40 min (or as indicated below). Standard medium contained 50 mM Tris-HCl buffer (pH 7.5), 0.5 mM EDTA, 0.05 mg/ml tRNA, and 0.02-0.06 mg/ml IgG. In some experiments, salt concentration and pH were varied (as indicated in figure legends). After the reaction, protein was removed from the reaction medium by phenol extraction (20 µl of phenol). RNA was pelleted by addition of 0.3 ml of 2% LiClO4 in acetone and centrifugation; the tRNA pellet was washed with 0.2 ml of acetone. After the second centrifugation, 6 µl of dye-containing urea was added to the mixture and samples were loaded onto the gel. tRNA hydrolysis products were analyzed by electrophoresis through 15% polyacrylamide gel in the presence of 8 M urea. Hydrolysis products were identified by autoradiography. Radioactivity of initial tRNA and its hydrolytic products was quantified by cutting the gel into pieces corresponding to these components and counting by the Cherenkov method.
Hydrolysis of tRNA by RNAse T1. Partial hydrolysis by RNAse T1 was performed in 10 µl of buffer (50 mM Tris-HCl, pH 7.5, containing 0.5 mM EDTA) and 1 µg carrier RNA as described [33]. The solution of tRNA was heated for 10 min at 55°C and cooled to 0°C; 5·10-3 units of RNAse T1 were added, incubated for 10 min at 55°C, cooled, and supplemented with 1 µl of ×20 Tris-borate buffer (1 M Tris-HBO4, pH 8.0, 20 mM EDTA), standard urea, and dye; samples were analyzed by electrophoresis.
Static Hydrolysis of tRNA with Imidazole. tRNA was hydrolyzed as described [34]; 2 µl of labeled RNA (1 µg), 3 µl of water, and 5 µl of 4 M imidazole buffer (pH 7.0) were mixed and incubated for 10 min at 90°C. The solution was cooled and 10 µl of 0.3 M sodium acetate were added; samples were prepared for electrophoresis by pelleting tRNA with lithium perchlorate solution as described above.
Reagents. Most reagents were from Merck and Euromedex (Germany); protein A-Sepharose was from Sigma (USA); Toyopearl HW-55 fine was from ToyoSoda (Japan); Triton X-100 was from Ferak (Germany); DNA of pBR-322 plasmid was from SybEnzyme (Novosibirsk, Russia); tRNA and enzymes for tRNA labeling were a gift from Dr. Kite (Strasbourg, France). All other reagents were of chemical grade.
RESULTS AND DISCUSSION
We assayed RNAse and DNAse activities of IgG Abs from the blood of healthy donors and patients with various forms of viral hepatitis. The immunoglobulins were purified by ammonium sulfate precipitation (50% saturation) and affinity chromatography on protein A-Sepharose. After Abs were absorbed on protein A-Sepharose, the column was washed with buffer containing Triton X-100 that destroys even relatively stable non-covalent protein complexes [18, 20]. It was demonstrated that washing the column with acidic buffer (pH 4.5-5.0) is a very efficient way of removing the contaminating proteins [15, 20]. The buffer elutes a small fraction of IgG with reduced affinity to protein A but does not elute the NA-hydrolyzing activity. Subsequent elution of Abs with more acidic buffer (pH 2.6) yielded DNA- and RNA-hydrolyzing Abs. The Abs from the blood of the healthy donors and patients with various diseases were used to compare their NA-hydrolyzing activities.
Activities of all Abs was tested by DNA hydrolysis during purification. It is important that relative NA-hydrolyzing activity of Abs (hydrolysis for 2 h) was highly variable depending on the patient and disease. A general system of quantitative evaluation of supercoiled DNA hydrolysis is rather difficult for a number of reasons. Sometimes, only single breaks were observed in a single DNA chain (Fig. 1, lanes 1-3). More active Abs hydrolyzed DNA forming products containing multiple breaks in both DNA chains and linear DNA (lanes 4-6). The most active Abs hydrolyzed DNA into oligonucleotides (lanes 7-10). Thus, a scale system was used to qualitatively evaluate arbitrary DNA-hydrolyzing activities of Abs; arbitrary units of Ab activity with grades from 0 to 10 were used as indicated in Fig. 1. Zero activity of Abs corresponds to lack of detectable changes in the amount of supercoiled (lower band of C1 lane) and relaxed DNA (upper band) after 2-h incubation with Abs.
Abs isolated from the blood of 50 healthy donors had no detectable activity (Fig. 1, lanes C2 and C3). A similar pattern was observed in Abs from the blood of patients with influenza, pneumonia, tuberculosis, tonsillitis, duodenal ulcer and some types of cancer (uterine tumors, breast cancer, intestinal, and gastric cancer). On the other hand, Abs from the blood of patients with all forms of viral hepatitis exhibited certain activity depending on the severity and duration of the disease and viral type (A, B, C, delta, or simultaneous infection with various types of the virus); the activity varied from 1 to 10 arbitrary units (Fig. 1). The clinical aspects of correlation of Ab activity with severity and duration of the infection, etc., will be described in a separate publication. However, it is important that the level of Ab activity was significant (at least 1 arbitrary unit) in the blood of all 49 patients with various forms of viral hepatitis.Fig. 1. Level of arbitrary activity of Abs from the blood of patients with viral hepatitis according to electrophoresis of hydrolytic products of pBR-322 plasmid supercoiled (lane C1, lower band) and relaxed DNA (lane C1, upper band) during its incubation with Abs (2 h at 37°C). Lanes: C1) DNA was incubated without Abs; C2, C3) DNA incubated with Abs from the blood of healthy donors; 1-10) Abs from the blood of 10 various patients with viral hepatitis. Numbers of the lanes 1-10 correspond to the scale values of arbitrary activity of Abs in DNA hydrolysis.
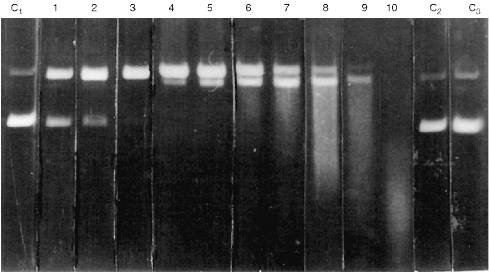
Considering the less active abzymes appearing in patients with other disorders, DNA was incubated with Abs for 12 h. This did not yield detectable NA-hydrolyzing activity of Abs from the blood of the healthy donors and patients with influenza and tonsillitis. In some other disorders (Table 1), detectable DNA hydrolysis was observed after 12-h incubation of the reaction medium; according to the scale presented in Fig. 1, it corresponds to 1-3 arbitrary units. Activity of Abs isolated from the blood of leukemia patients varied from 2 to 5 arbitrary units (12-h incubation). The data on activity of Abs in various diseases are summarized in Table 1. It should be noted that in all cases, apart from hepatitis, the arbitrary units of activity corresponding to the distribution of DNA hydrolysis products analyzed by electrophoresis (Fig. 1) are presented for 12-h incubation and in case of hepatitis, these correspond to 2-h incubation.
TABLE 1. Arbitrary DNA-Hydrolyzing Activity
of IgG Antibodies from the Blood of Patients with Various Disorders
A similar pattern was observed in assay of RNA-hydrolyzing activities of Abs from the blood of the healthy subjects and patients with various diseases. Abs from the blood of patients with acute forms of various types of viral hepatitis had maximal RNA-hydrolyzing activity. The data indicate that in hepatitis patients (and patients with autoimmune disorders), Abs that very efficiently cleave NA are accumulated. However, such Abs were not detected in patients with another viral disease, influenza, and some other disorders associated with inflammation.
The data indicate that NA hydrolysis is an intrinsic function attributable to Abs. However, when studying abzymes, it is very difficult to prove that the chemical reactions are catalyzed by the immunoglobulins but not by a contaminating nucleases which can be co-purified with Abs. To prove that the catalytic function is an intrinsic property of the IgG, the enzymatic activity of Abs from the blood of patients with viral hepatitis was additionally investigated.
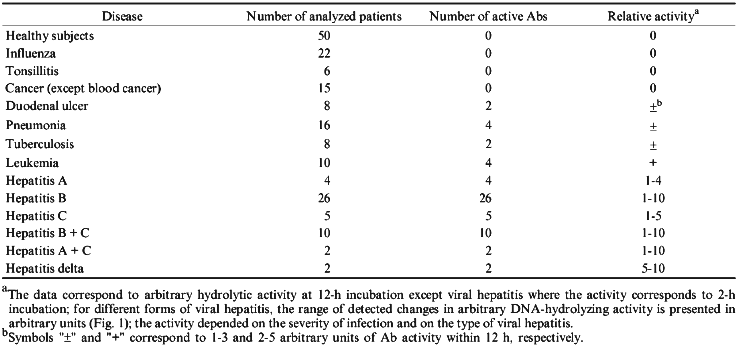
Following the purification of Abs by affinity chromatography on protein A-Sepharose, IgG were additionally purified by DEAE-cellulose chromatography, gel filtration on a Toyopearl HW-55 column, and affinity chromatography on DNA-cellulose. After all these purification stages, Abs still possessed DNA- and RNA-hydrolyzing activities (Fig. 2).
The silver staining of Abs electrophoresed in the absence of 2-mercaptoethanol reveals a single protein corresponding to 150 kD IgG oligomer. Under dissociating conditions of electrophoresis (with 2-mercaptoethanol), two protein bands are stained corresponding to the molecular weights of the light and heavy Ab chains. Other silver-stained protein bands were not detected (Fig. 3). Identity of these bands to the light and heavy IgG chains was additionally supported by immunoanalysis; Abs were positively stained with polyclonal anti-IgG and their subunits reacted with Abs to light and heavy IgG chains.Fig. 2. Electrophoretic analysis of pBR-322 plasmid DNA hydrolysis by Abs from the blood of patients with hepatitis after various stages of their purification. Lanes: PS) protein A-Sepharose; GF) gel filtration; AF) affinity chromatography on DNA cellulose; pH) gel filtration in acidic buffer (pH shock); F1) preparation of homogenous Fab fragments; a-IgG) Abs after addition of IgG Sepharose; C1) DNA incubated without Abs; C2) Abs from the blood of an influenza patient. Mobility of relaxed DNA is identical with that of DNA containing a break in one of the strands.
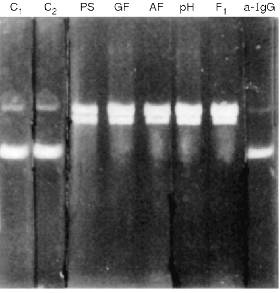
Additional evidence supporting the assumption that IgG but not possible contamination with blood nucleases are responsible for DNA- and RNA-hydrolyzing activities were obtained.Fig. 3. SDS electrophoresis of homogenous IgG abzymes (lanes 2 and 3) and Fab fragment (1) without (1 and 2) and with 2-mercaptoethanol (3). Lane C, protein markers with known molecular weight.

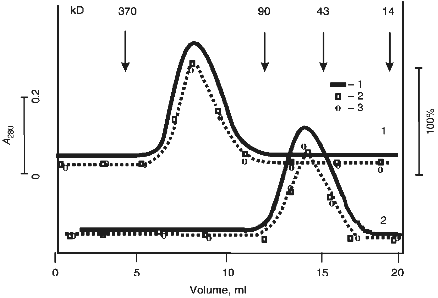
The protein absorption profile of gel filtration in acidic buffer (after incubation of IgG Abs in this buffer at pH 2.6; acidic shock) coincided with the profiles of DNA- and RNA-hydrolyzing activities (Fig. 4). Similar data were obtained in case of Fab fragments of Abs.
Incubation of Abs with anti-IgG Sepharose completely removed all hydrolyzing activities from the solution (Fig. 2).Fig. 4. Gel filtration of Abs (curve 1) and their Fab fragments (curve 2) on a Toyopearl HW-55 column equilibrated with 50 mM glycine-HCl buffer (pH 2.4) containing 0.3 M NaCl after preincubation of Abs and their fragments (IgG were purified by DNA-cellulose chromatography) in the same buffer: 1) protein absorption at 280 nm; 2) relative activity of Abs in hydrolysis of pBR-322 DNA; 3) [gamma-32P]r(pA)10. The maximal relative activity in hydrolysis of each of the substrates (DNA and [32P]r(pA)10) was taken as 100%.
Electrophoretically homogenous preparation of Fab fragments of Abs (Figs. 3 and 4) exhibited DNA- and RNA-hydrolyzing activities (Figs. 2 and 5). The rates of hydrolysis of DNA, ribooligonucleotides, tRNA, and CCMP by Fab fragments were only 1.3-2-fold lower versus the initial Abs.
Five ribonuclease with variable affinity to phosphocellulose have been described in human blood [35]. Nevertheless, the substrate specificity of all five ribonucleases is similar to RNAse A whose preferred substrates are poly(C) and poly(U); all enzymes very poorly cleave poly(A) [35-40]. All five ribonucleases from human blood preferentially hydrolyze poly(C) (100%) and the efficiency of poly(U) hydrolysis varies from 3 to 16%; significant hydrolysis of poly(A) was not demonstrated. Moreover, all five ribonucleases of blood serum are 1.7-2.7-fold activated by 50-100 mM NaCl or 10 mM MgCl2.Fig. 5. Electrophoretic analysis of [32P]tRNAPhe hydrolysis by RNAse A (a) and Abs from the blood of hepatitis patients (b) under various conditions. Lanes: 1 (a) and 2-6 (b) (Ab preparations from 5 donors), hydrolysis without salts; 2 (a) and 10-12 (b), hydrolysis in the presence of 100 mM NaCl; 3 (a) and 7-9 (b), hydrolysis in the presence of 10 mM MgCl2 (corresponds to lanes 4-6); 4 (a) and 13 (b), hydrolysis in the presence of 100 mM NaCl and 10 mM MgCl2 (corresponds to lane 4); 5 (a), partial hydrolysis of tRNA by RNAse T1; 1 (b), Fab fragments corresponding to Abs in lane 4.
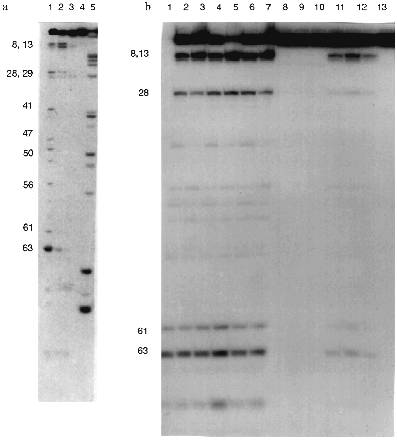
Unlike the nucleases of human blood, Abs of hepatitis patients preferentially hydrolyze poly(A), show little hydrolysis of poly(U), and are essentially inactive towards poly(C) (Table 2). Their cCMP-hydrolyzing activity is about 4-5% of that of RNAse A. Moreover, RNAse activity of hepatitis Abs (unlike human blood RNAses) was almost completely suppressed in the presence of 100 mM NaCl or 10 mM MgCl2 (Fig. 5). Relative activities of Abs of hepatitis patients in hydrolysis of various homopolymers were different from those of ELE Abs (Table 2) and from those in patients with PA and AIT [15].
TABLE 2. Relative Specific Activities of
RNAse A, Human Blood Nucleases, and Abs from the Blood of Hepatitis
Patients in Hydrolysis of Various Polynucleotides and
cCMPa
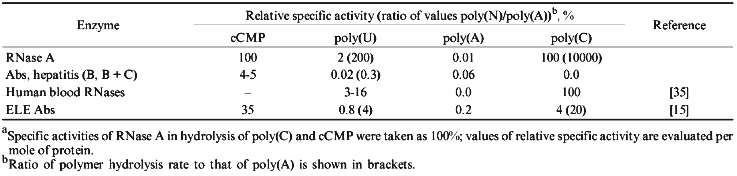
Thus, analysis of our data and results of other authors suggests that considering the relative hydrolysis rates and substrate specificity of cleavage of various homopolynucleotides, Abs of hepatitis patients are significantly different from human blood ribonucleases and from pancreatic RNAse A.
The next stage in characterization of the substrate specificity of abzymes included their comparison with RNAse A in hydrolysis of yeast tRNAPhe (the structure of yeast tRNAPhe is shown in Fig. 6). Hydrolysis of 3´-labeled [32P]tRNAPhe by RNAse A, Abs, and Fab fragments is shown in Fig. 5.
The following conditions and protein concentration (according to Cherenkov radioactivity counting) were selected for such comparison: the band corresponding to the initial tRNA after its hydrolysis for 40 min should contain about 70-80% of the initial tRNA. The sites of tRNA hydrolysis by RNAse and Abs were determined using static hydrolysis of tRNA by imidazole and partial cleavage by RNAse T1.Fig. 6. Structure of yeast tRNAPhe. Arrows indicate the main cleavage positions by RNAse A and Abs in the absence of salts; tRNA break points and cleavage positions corresponding to both RNAse and Abs are emphasized by big numbers near the arrows.
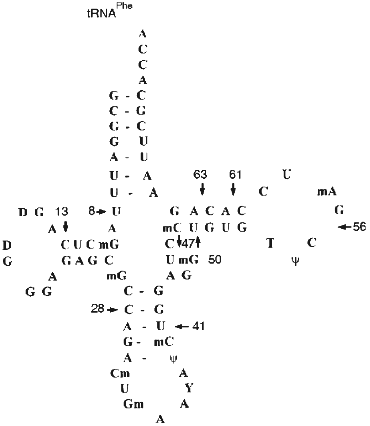
The data of Fig. 5a (lane 1) indicate that RNAse A hydrolyses 3´-[32P]tRNAPhe without salts at sites corresponding to positions 63 (between nucleotides 63 and 64), 8, 13, 28, 41, 47, 50, 56, and 61. Under similar conditions, Abs from the blood of 5 patients (Fig. 5b, lanes 2-6) and Fab fragments of Abs (lane 1 (b)) have identical cleavage pattern. In all cases the most intensive hydrolysis occurred at positions 8, 63, 61, 13, and 28. Unlike RNAse A, hepatitis Abs preferentially hydrolyze tRNA at position 8 but not 63 and do not hydrolyze at positions 41, 47, 50, 56, and other sites between positions 28 and 61. Addition of 100 mM NaCl inhibits RNAse A (Fig. 5a, lane 2) and Abs (Fig. 5b, lanes 10-12). However, general inhibition of RNAse is associated with its activation in tRNA hydrolysis at positions 8 and 13; inhibition of Abs results in lowering the hydrolytic efficiency in all tRNA positions including positions 8 and 13 to the same extent.
In the presence of magnesium ions, RNAse activity is significantly inhibited although not by 100% (Fig. 5a, lane 3). In the presence of magnesium ions, Abs cannot hydrolyze tRNA (Fig. 5b, lanes 7-9). Simultaneous addition of NaCl and MgCl2 completely inhibits RNAse A and Abs (Fig. 5a, lane 4; Fig. 5b, lane 13). Thus, assuming tRNA hydrolysis at position 63 as a standard point of evaluation of relative intensities of tRNA hydrolysis, relative rates of tRNA hydrolysis by RNAse A and hepatitis Abs at various positions significantly differ. Moreover, RNAse A activity is not affected by preincubation for 1 h at 70°C whereas activity of Abs is suppressed by 90-95%.
Thus, the data indicate that Abs from the blood of patients with viral hepatitis can hydrolyze DNA and RNA.
It was noted above that we have previously compared specificity of Abs from the blood of patients with ELE, PA, and AIT. It is important that unlike RNAse A and hepatitis Abs, magnesium ions stimulate unique specificity of these Abs including cleavage of tRNAPhe and tRNALys at double strand regions of tRNA. Their specificity is somewhat similar to nuclease V1 from cobra venom [41]; however, specificity of Abs and nuclease V1 are not completely identical. Moreover, ELE, PA, and AIT Abs are significantly different in hydrolysis of homopolynucleotides and tRNA.
Specificity of Abs from breast milk of women in hydrolysis of various polynucleotides and tRNA also significantly differs from specificity of RNAses from human blood, milk, and tissues [20, 35-40]. Thus, RNA- and, possibly, DNA-hydrolyzing activities of Abs can be used to develop the methods of diagnostics of various autoimmune and viral diseases.
This work was supported by the Russian Foundation of Basic Research (grant No. 95-04-12950) and State Scientific and Technical Program "New Methods in Bioengineering" (grant No. 03.0004H-364).
LITERATURE CITED
1. Tramontano, A., Janda, K. D., and Lerner, R. A.
(1986) Science, 234, 1566-1569.
2Lerner, R. A., and Tramontano, A. (1981) Trends Biochem. Sci.,
12, 427-438.
3. Lerner, R. A., Benkovic, S. J., and Shultz, P. J.
(1991) Science, 252, 659-667.
4. Benkovic, S. J. (1992) Annu. Rev. Biochem.,
61, 29-54.
5. Hilvert, D. (1992) Pure Appl. Chem.,
64, 1103-1113.
6. Suzuki, H. (994) J. Biochem.,
15, 623-628.
7. Paul, S., Volle, D. J., Beach, C. M., Johnson, D.
R., Powell, M. J., and Massey, R. J. (1989) Science,
244, 1158-1161.
8. Li, L., Paul, S., Tyutyulkova, S., Kazathikine, M.
D., and Kaveil, S. (1995) J. Immunol., 154,
3328-3332.
9. Shuster, A. M., Gololobov, G. V., Kvashuk, O. A.,
Bogomolova, A. E., Smirnov, I. V., and Gabibov, A. G. (1992)
Science, 256, 665-667.
10. Gololobov, G. V., Chernova, E. A., Schourov, D.
V., Smirnov, I. V., Kudelina, I. A., and Gabibov, A. G. (1995) Proc.
Natl. Acad. Sci. USA, 92, 254-257.
11. Schourov, D. V., Makarevich, O. I., Lopaeva, O.
A., Buneva, V. N., Nevinsky, G. A., and Gabibov, A. G. (1994) Dokl.
Ros. Akad. Nauk, 337, 407-410.
12. Buneva, V. N., Andrievskaya, O. A., Romannikova,
I. V., Gololobov, G. V., Yadav, R. P., Yamkovoi, V. I., and Nevinsky,
G. A. (1994) Mol. Biol. (Moscow), 28, 738-743.
13. Vlassov, A. V., Andrievskaya, O. A., Kanyshkova,
T. G., Baranovsky, A. G., Naumov, V. A., Breusov, A. A., Giege, R., and
Nevisky, G. A. (1997) Biochemistry (Moscow), 62, 559-565
(Russ.).
14. Andrievskaya, O. A., Kanyshkova, T. G.,
Yamkovoi, V. I., Buneva, V. N., and Nevinsky, G. A. (1997) Dokl.
Ros. Akad. Nauk, in press.
15. Vlassov, A. V., Baranovsky, A. G., Kanyshkova,
T. G., Prints, A. V., Zabara, V. G., Naumov, V. A., Breusov, A. A.,
Giege, R., Buneva, V. N., and Nevinsky, G. A. (1997) Mol. Biol.
(Moscow), 31, 1117-1127.
16. Buneva, V. N., Kanyshkova, T. G., Semenov, D.
V., and Nevinsky, G. A. (1996) Proc. 2-nd Int. Conf. "Catalytic
Antibodies and Antibody Engineering", Chantilly, France,
October 14-18, p. 11.
17. Buneva, V. N., Andrievskaya, O. A., Kanyshkova,
T. G., Vlassov, A. V., Baranovsky, A. G., and Nevinsky, G. A. (1996)
Proc. 4-th Int. Meet. "Ribonucleases: Chemistry, Biology,
Biotechnology", Groningen, Netherlands, July 14-18, S7P7.
18. Kit, Yu. Ya., Semenov, D. V., and Nevinsky, G.
A. (1995) Mol. Biol. (Moscow), 29, 519-526.
19. Kit, Yu. Ya., Semenov, D. V., and Nevinsky, G.
A. (1996) Bioch. Mol. Biol. Int., 39, 521-527.
20. Kanyshkova, T. G., Semenov, D. V., Vlassov, A.
V., Khlimankov, D. Yu., Baranovsky, A. G., Shipitsin, M. V., Yamkovoi,
V. I., Buneva, V. N., and Nevinsky, G. A. (1997) Mol. Biol.
(Moscow), 31, 1086-1096.
21. Shoenfeld, Y., Ben-Ehuda, O., Messinger, J.,
Bentwithc, Z., Rauch, J., Isenberg, D. I., and Gado, N. (1988)
Immunol. Lett., 17, 285-291.
22. Coll, J., Palazon, J., Yazbeck, H., Gutierrez,
J., Aubo, C., Benito, P., Jagiello, P., Maldyk, H., Marrugat, J., and
Anglada, J. (1995) Clin. Rheumatol., 14, 451-457.
23. Gololobov, G. V., Mikhalap, S. Y., Starov, A.
V., and Gabibov, A. G. (1993) Proc. 1-st Int. Conf. "Catalytic
Antibodies and Antibody Engineering", Moscow, Russia, June
6-11, p. 25.
24. Bigazzi, P. E. (1983) in Mechanisms of
Immunology (Cohen, S., Wozd, P. F., and MacKlaski, R. T., eds.)
[Russian translation], Meditsina, Moscow, pp. 181-206.
25. Laemmli, U. K. (1970) Nature,
227, 680-685.
26. Merril, C. R., Goldman, D., and Van Keuren, M.
L. (1982) Electrophiresis, 33, 17-26.
27. Towbin, H., Staehelin, T., and Gordon, J.
(1976) Proc. Natl. Acad. Sci. USA, 9, 4350-4356.
28. Harlow, E., and Lane, D. (1988) Antibodies: A
Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring
Harbor, NY, pp. 630-631.
29. Lee, J. S., Dombrovskii, D. F., and Mosmann, T.
(1988) Biochemistry, 21, 4940-4947.
30. Crook, E. M., Mathias, A. P., and Rabin, B. R.
(1960) Biochem. J., 74, 234-238.
31. Jrie, M., Mikami, F., Momna, K., Ohgi, K.,
Watanabe, H., Yamaguchi, R., and Nagase, H. (1984) J. Biochem.,
96, 89-96.
32. Milligan, J. F., Groebe, D. R., Witherell, G.
W., and Uhlenbeck, O. C. (1987) Nucleic Acids Res., 15,
8783-8798.
33. Vlassov, V. V., Giege, R., and Ebel, J.-P.
(1981) Eur. J. Biochem., 119, 51-59.
34. Vlassov, V. V., Zuber, G., Felden, B., Behr,
J.-P., and Giege, R. (1995) Nucleic Acids Res., 23,
3161-3167.
35. Akagi, A., Merai, K., Hirao, N., and Yamanaka,
M. (1976) Biochim. Biophys. Acta, 442, 368-378.
36. Shapot, V. S. (1968) Nucleases [Russian
translation], Meditsina, Moscow, pp. 1-162.
37. Zimmerman, S. B., and Sandeen, G. (1964)
Analyt. Biochem., 10, 444-449.
38. Watt, C. W., and Houck, J. C. (1964) Biochim.
Biophys. Acta, 89, 90-94.
39. Imura, B. N., Irie, M., and Ukita, T. (1965)
J. Biochem., 58, 264-272.
40. Ramaswamy, H., Swamy, Ch. V. B., and Das, M. R.
(1993) J. Biol. Chem., 268, 4181-4187.
41. Butorin, A. S., Remy, P., Ebel, J. P., and
Vassilenko, S. K. (1982) Eur. J. Biochem., 121,
587-595.