Telomere-Binding Protein from the Nuclear Envelope of Oocytes of the Frog Rana temporaria
E. A. Bugaeva1 and O. I. Podgornaya1,2
1Institute of Cytology, Russian Academy of Sciences, Tikhoretskii pr. 4, St. Petersburg, 194064 Russia; fax: (812) 520-9703; E-mail: podg@osint.stud.pu.ru2To whom correspondence should be addressed.
Submitted July 7, 1997.
The identification of telomere-binding protein on the nuclear envelope (NE) of the frog oocyte is reported here. Nuclei and the NE were isolated manually and extracted, and the extracts were used to search for DNA--protein specific interactions. Fragment of the Tetrahymena telomere sequence (T2G4)130 from the plasmid YAC4 were used as a labeled probe. DNA--protein complexes revealed by gel shift assay were cut out of the gel and injected into a guinea pig. The antibodies obtained have common antigenic determinants with keratins; this was shown on Western-blot under conditions of competitive binding. Antibodies were purified on an affinity column with keratins attached and used in the following work. Antibodies recognize one protein with molecular weight 70 kD among the NE proteins; no signal was obtained to proteins isolated from the inner part of the oocyte, but two polypeptides of 70 and 120 kD were detected in the proteins from frog liver nuclei. The 70-kD protein is situated on the NE in a distinct pattern that looks like a network, probably of double-dots as was shown by immunofluorescence of the NE. Electron-microscope immunogold staining showed that the protein is localized in the outer surface of the oocyte NE within cup-like structures attached to the membrane. Combined in situ hybridization using the mammalian telomeric DNA sequence (T2AG3)135 and immunocytochemistry using antibodies showed them to be colocalized in the frog blood cells, so telomere sequence coexists with the protein in the interphase nuclei. Most of the telomeres are fused in highly differentiated blood cells though a double dot signal is visible. The signal appears to be localized in the outer part of the nuclei. The existence of membrane telomere-binding protein allows a discussion of the attachment of the telemeters to the membrane.
KEY WORDS: telomere-binding protein, oocyte, nuclear membrane.
Whether telomeres are bound to the membrane or not is an old question. Rabl was the first to observe in 1885 [1] that the ends of prophase chromosomes seemed to occupy a peripheral position at one end of the nuclei while the centromeres were at the other end. The model based on this observation supposes that there is a mechanism of attachment of these sequences to the protein layers tightly bound to the membrane if not to the membrane itself. The research of more than one hundred years produced a large amount of controversial experimental material but did not give an answer to this question [2].
There is no doubt that telomeres are attached to the nuclear membrane in one case--in the middle meiotic stages during gametogenesis. The prophase stage of the first meiotic division is durable, several months in the case of frog oocytes. It is divided into several stages, i.e., leptotene, zygotene, pachytene, diplotene, and diakinesis, according to the visible position of chromatin in the process of homologous chromosome pairing. The chromosomes are connected all along by the synaptonemal complex when the pairing is finished--at zygotene and pachytene--causing the chromosomes to adopt the configuration termed the "bouquet" formation. All the telomeres in the bouquet are connected and associated with the nuclear membrane. At the next diplotene stage the chromosomes undergo compaction and lose their connection with the nuclear envelope. There is a special mechanism of meiotic inactivation of the chromatin in the case of frog oocytes, namely, chromosomes assemble in the middle of the nucleus and become covered with a special protein structure, the capsule karyosphere [3].
Nuclear envelope (NE) of the frog oocyte nuclei (stage 6 according to [4]) was chosen as a source of protein for the search of a telomere-binding protein because it has a number of the unique properties. Oocyte nuclei are large, about 1000 µm in size; these can be isolated manually. Common biochemical methods of membrane isolation damage or even destroy the protein membrane-attached layers [5]. Conditions for shrinkage of the inner part of the oocyte nucleus are known; after this it is also possible to isolate the NE under the microscope by hand. As a result the entire chromatin telomere-binding proteins cannot interfere with the membrane proteins because the chromatin is removed from the NE preparations in the capsule karyosphere. Chromosomes are not attached to the NE at the 6th stage when isolation is done, but most of the metabolic processes in the oocyte are slow and there was the possibility that we could catch the biochemical "memory" of the previous stages of bouquet formation.
The protein which we found is an integral component of the thick membrane--the nuclear envelope. We term it MTBP (Membrane Telomere-Binding Protein) for this paper. It is probably identical to the already described Tbf1 [6]. To known features of this protein the present work adds information on its membrane localization in the oocyte nuclei and its surface localization in fully differentiated somatic cells. We hope that in the future immunofluorescence experiments with antibodies (AB) obtained against MTBP will show that in other somatic cells MTBP is also associated with the membrane and therefore in the majority of cells at most stages of the cell cycle telomeres are attached to the membrane.
MATERIALS AND METHODS
Isolation of Protein Preparations from Oocytes. Frogs Rana temporaria were collected in autumn and kept at 4°C during winter. In March the oocytes in the ovary developed to diplotene stage 6 [4].
Oocytes 1200-1400 µm in size were used. The nuclei were isolated manually under an MBS-1 microscope in TBS buffer (10 mM Tris-HCl, pH 7.2, 83 mM KCl, 17 mM NaCl) [7]. Up to 10 mM MgCl2 was added to the same buffer to isolate nuclear envelopes. This induces shrinkage of the nucleoplasm and its separation from the membrane. This allowed isolation of the nuclear envelopes with needles. Separated nuclear envelopes and the inner parts were stored at -70°C in an isolation solution or used immediately for protein extraction. All the buffers and protein extraction solutions contained 5 mM PMSF and 2.5 mM DTT. Envelopes were extracted sequentially with solutions I (10 mM Tris-HCl, pH 7.2, 0.14 M NaCl, 0.1% Triton X-100) and II (10 mM Tris-HCl, pH 7.2, 0.8 M KCl, 1% Triton X-100). The protein was extracted at 4°C for 1-2 h with homogenization in a microhomogenizer. The extract was clarified by centrifugation for 15 min at 10,000 rpm. Usually 2000 NE were extracted in 100 µl of solution for an experiment. The protein concentration in the resulting extracts was 0.1-0.3 mg/ml according to electrophoresis and the Bradford method [8].
Frog Liver Nuclei Isolation. The same method as for rat and mouse liver nuclei was used for the frog liver nuclei isolation [5]. Liver tissue was homogenized in 10 volumes of the solution containing 0.32 M sucrose, 25 mM Tris-HCl, pH 7.5, 5 mM MgCl2, and 1 mM PMSF. After centrifugation at 1000 rpm raw nuclei were loaded on 2 M sucrose cushion and pelleted at 100,000g for 40 min. The purity of the preparations were monitored using a microscope; pure nuclei were used for electrophoresis.
Plasmid Isolation and Band Shift Assays. Plasmid YAC4 contains a Tel:His3:Tel construct [9]. Two Tetrahymena (T2G4)130 telomere inserts around 800 bp each can be excised with a BamHI/HindIII digest. The plasmid was the gift of Dr. V. Larionov (Institute of Cytology, Russian Academy of Sciences).
Plasmid pSXneo containing (T2AG3)135 sequence (the vertebrate telomere) was a gift from Dr. R. Bayne (HGU MRC, Western General Hospital, Edinburgh). Telomere repeats from plasmid pSXneo, of 800 bp in length, could be excised by BamHI/BglII double digests. Inserts were isolated from agarose gel, end-labeled with [alpha-32P]dCTP using the Klenow fragment and used in band shift assays [10]. For in situ hybridization the fragment was first isolated from a gel and then labeled with biotin-16-dUTP; in a random primer reaction DNA was separated from nucleotides on a column and the effectiveness of labeling was tested according to the standard solutions of biotinylated phage lambda DNA.
Band shift assays basically followed the procedure of Strauss and Varshavsky [11]. A free fragment runs with xylene cyanole in 3.5% polyacrylamide gels and electrophoresis continues for 3 h at 25 mA/cm2. Total genomic E. coli DNA ultrasonicated to 1 kb was used as a competitor. The incubation mixture for the band shift assay used contained 1 mM MgCl2, though later it was found that binding is not Mg-dependent. The concentration of labeled probe, unlabelled competitor, and proteins used in band shift assays is given in the figure legends. Unless stated otherwise, ultrasonicated E. coli DNA was the competitor DNA.
Preparation of Antibodies and Their Affinity Purification. After determining optimal conditions for band shifts (Fig. 1a, lanes 3 and 7) a number of preparative gels (18 × 24 cm) were run. The gels were dried between cellophane and exposed on X-ray film (Kodak XAR-5). Retarding complexes were cut out according to the autoradiograph, powdered in liquid nitrogen (cellophane was removed at this stage) and injected into guinea pig intraepidermally in physiological solution with complete Freund's adjuvant at 1:1 ratio. Two male guinea pigs, each three-months-old with approximate weight of 300 g, were immunized. Complexes from one gel (extracts from 2000 NE loaded) were used for each booster. The injection was repeated after 14 days, and blood was obtained from the heart one week after the last injection. Serum was obtained and stored at 4°C in the presence of sodium aside.
AB from the serum was affinity purified on human keratins after the stage when common antigenic determinants between MTBP and keratins had been found (Fig. 3b). Affinity purification was performed on human skin keratins. For this 30-70 mg of tissue (dead cells from around the nails) were boiled in 0.5 ml of 10% SDS and 10% beta-mercaptoethanol. Then 0.5 ml of 7.5% NaHCO3 was added. The keratins were bound to 100 mg of dry BrCN-Sepharose overnight at 4°C with shaking. The Sepharose was packed into a column, washed with 7.5% NaHCO3, and blocked with Laemmli electrophoresis buffer (×10) [12]. The column then was first washed with TBS, 1.5 ml of serum was loaded for 30 min with shaking, then the column was washed with 3 volumes of TBS and 3 volumes of TBS with 0.5 M NaCl and again with TBS to remove nonspecific binding. Bound proteins were eluted with 0.1 M glycine, pH 2.0. The pH of the eluate was immediately adjusted to pH 7.0 with Tris-base. As a rule, 1 ml of eluate was obtained from the column. Affinity-purified antibodies were tested by Western-blot.
Monoclonal antibodies against vimentin (Boehringer Mannheim, Germany, cat. No. 1112457) in working dilution 1:100 were also used for Western-blots. Antibodies against Xenopus lamins were a kind gift of Dr. G. Krohne (Institute of Cell and Tumor Biology, Heidelberg, Germany). AB 167-1 (not species-specific, not lamin-specific) were used for Western-blot and AB 155-8 with specificity to LII and LIII of Xenopus laevis for immunofluorescence.
SDS-PAGE in 12% and 10% gels was done according to Laemmli [12]. Protein A labelled with 125I and biotin-conjugated II AB (goat anti guinea pig, Sigma, USA) with the following staining by streptavidin-alkaline phosphatase were used in order to reveal the I AB.
Immunogold Electron Microscopy. To define the localization of the MTBP on the membrane, electron microscopy was done with affinity-purified AB. Oocytes at stage 6 [4] were fixed in 4% para-formaldehyde (PFA) in TBS, pH 7.4, for 24 h at 4°C, then in 0.1% glutaraldehyde in PFA-TBS at room temperature for 1 h. After washing in TBS and alcohol dehydration the oocytes were imbedded in LR WHITE resin (London Resin Company Ltd., England). Ultrathin sections (70 nm) were obtained on an ultratome from Reichert-Yung (Austria). The sections were loaded on grids and incubated for 15 min in each of the following: TBS with 0.02 M glycine, then TBS with 0.5% gelatin for blocking. Incubation with the I AB was carried out for 1.5 h at 37°C in working dilution from 1:500 to 1:4000. For antibody detection an electron-dense labeled protein A conjugated with colloidal gold with particles of 10 nm in diameter was used (F10). Grids were screened on JEM-7A and JEM-100C (Japan) electron microscopes at increasing current (voltage) 80 kV.
Immunofluorescence. Blood from 1-2 frogs was collected into glass tubes treated with heparin. The tubes were stored in a refrigerator at 4°C for 0.5 h, and the supernatant was used for cell spread on slides. In this way the preparation was enriched with lymphocytes. The slides were fixed in 4% formaldehyde in TBS or KCM (for KCM solution see the next section) solutions and then proceeded as described below for the NE.
The procedure for NE spread is based on the method described by Callan [13] with some modifications. The nuclei were washed from the yolk by pipetting and NE separated as described above. NE were loaded into chambers attached to slides [14] and containing TBS. The nuclei were centrifuged for 15 min at 3700-4000 rpm, then the attached envelopes were sequentially washed with 70% alcohol followed by phosphate buffer, pH 7.2, and processed for immunofluorescence as follows: I AB, II AB, and streptavidin--FITC conjugate (for 1 h each with washing in TBS in between). Opton (Germany) and Zeiss Axioplan (Germany) fluorescence microscopes with automatic computer processing of images with software of Digital Scientific (Cambridge, England) [15] were used for viewing and taking pictures. In the latter case the cells were also stained with DAPI.
Combined Immunocytochemistry and in situ Hybridization. This was carried out basically following the procedure already described [16]. Frog blood cells spread on a slide were fixed in 4% formaldehyde in KCM solution (135 mM KCl, 20 mM NaCl, 0.5 mM EDTA, 0.1% Triton X-100, 10 mM Tris-HCl, pH 8.0) and stored in this solution before use [17]. All antibody reactions were carried out in KCM with 0.5-5% BSA as the blocking agent. Antibody reactions were performed in a moist chamber at room temperature for at least 1 h. Working dilution for affinity-purified antibody or serum are indicated in the figure legends. The primary signal was detected with the FITC-conjugated goat anti-guinea pig antibody (Sigma, USA). Mammalian telomeric DNA sequences (T2AG3)135 from the plasmid pSXneo (~500 ng) were labeled with biotin--dUTP (Boehringer Mannheim) by random prime nick-translation under standard conditions. For in situ hybridization the chromosomal DNA was denatured in 70% formamide/×2 SSC at 70°C for 3 min followed by dehydration through ice-cold 70, 90, and 100% alcohol. Probe DNA (~80 ng) was denatured by heating for 5 min, ice-cooled for 3 min, and hybridized overnight in 50% formamide/×2 SSC at 37°C with ultrasonicated salmon sperm DNA as carrier. Then the slides were washed in ×2 SSC at 50°C. Texas Red--avidin D (Vector Labs., USA) was used to detect the labeled probe on the chromosomal DNA. Cells were finally stained with DAPI. Images were taken with a Zeiss Axioplan fluorescence microscope equipped with software from Digital Scientific.
RESULTS
Identification of the DNA--Protein Complexes. Using the nitrocellulose filter binding assay, it was shown in our previous work that NE preparations have a clear preference in telomere fragment binding in contrast to the inner part of the oocyte nuclei [18]. We tried to extract telomere-binding activity with different solutions. Solutions of different ionic strength from physiological up to 1 M NaCl and different amounts of detergents, for example, Triton X-100 up to 1%, were used. We could not fully extract the activity even after sequential treatment of NE with increasingly strong solutions as shown with the same filter assay [18]. Proteins from some extracts did react with a telomere fragment on band shift assay in spite of incomplete extraction and gave visible specific complexes (Fig. 1a). There are multiple complexes in the experiment shown in Fig. 1a and such a picture is characteristic for this binding. The presence of the detergent in the extract solution is more important for the successful extraction of the activity than the increase in ionic strength. The low solubility of the protein(s) responsible for telomere-binding activity for the first time gave us the idea that it may resemble proteins of the intermediate filament class.
The protein composition of the NE is not so rich as that of the inner part of the nuclei. There are almost no polypeptides with lower molecular weight than 30 kD in the NE. The pattern of the polypeptides of the extracts was also analyzed, but despite the presence of a few proteins we were unable to identify the polypeptide responsible for the telomere-binding activity [18].Fig. 1. Gel retardation of the (T2G4)130--protein complexes (a) and testing of the serum raised against this complexes (b). Per each lane, 1 ng of the labeled fragment was used (E. coli DNA used as a competitor). a: 1) Free fragment; 2-5) 5 µl of the NE extract I added without competitor and with 10-, 20-, and 50-fold excess of the competitor, respectively; 6-8) 3 µl of the NE extract II added without competitor and with 20- and 50-fold excess of the competitor, respectively. b) Samples (1-3) contained 1 ng of labeled DNA, 7 µl of NE extract II, and 60-fold excess of the competitor. In addition, pre-immune and immune serum (both at dilution 1:200) were added to samples (2) and (3), respectively; 4) free fragment; 5) markers. On the right, sizes of the restricted fragments in kb. Immune serum causes hypershift of the complexes.
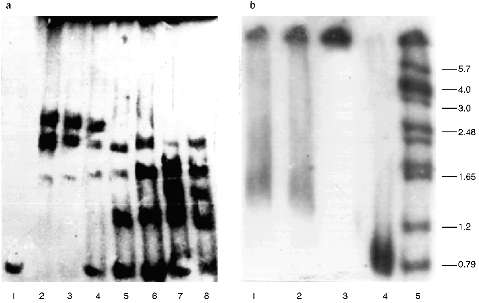
Thus, the telomere--protein complexes obtained on preparative gels under optimal conditions which were found in the analytical variant (Fig. 1a) were cut out and injected into a guinea pig.
Characterization of the Antibodies Obtained. The immune serum obtained decreases the mobility of the complexes, i.e., it causes hypershift, while the pre-immune serum added to the mixture in the same amount alters the retardation picture but does not destroy the complexes (Fig. 1b).
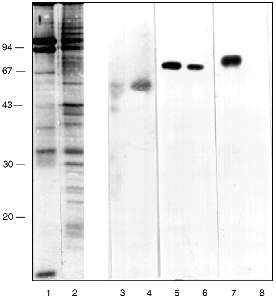
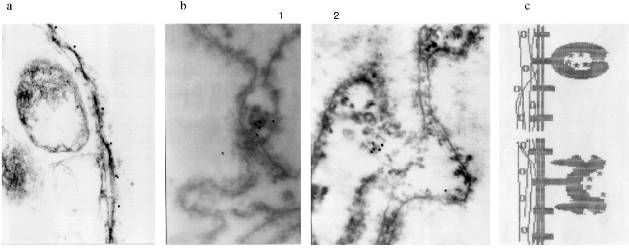
In the first attempts to test the serum it was found that antibodies (AB) of the immune serum recognize a polypeptide of around 70 kD in the preparations from frog, mouse, and human cells. Lamins with molecular weight 65-70 kD are known to be the main DNA-binding membrane proteins [19]. Vimentin with molecular weight 57 kD is an intermediate filament protein and it is known as a DNA-binding protein of the nuclear matrix [20] and at the same time it is closely associated with nuclear membrane [21]. Western-blot with the appropriate AB shows that lamin exists in the inner part of the nuclei as well as in the NE. Its relative amount is higher in the NE (Fig. 2, lanes 5 and 6). Vimentin was also found in both nuclear compartments with the opposite ratio (Fig. 2, lanes 3 and 4). Our own AB obtained in guinea pig against (T2G4)130--protein complexes reveal a 70 kD polypeptide among the NE proteins and nothing in the inner part of the nuclei (Fig. 2, lanes 7 and 8). The polypeptide with molecular weight 70 kD from the NE does not comigrate with lamin or vimentin (Fig. 2).
We checked the supposition that our AB recognize also antigenic determinants of keratins in the following way. Gel for electrophoresis was loaded with frog protein preparations in several wells and with keratins in several others. After electrophoresis half of the gel was stained (Fig. 3a) and the proteins from the other half were transferred to a nitrocellulose filter. Part of the filter with the keratins on it was incubated in the immune serum which contained AB against MTBP, washed, and then co-incubated overnight together with the part of the filter which contained frog proteins transferred. We supposed that AB would attach to the keratins on the first incubation, but if the affinity of the AB to the frog proteins were higher, the AB would be competed out from the keratins to the frog proteins and we should be able to observe staining. In other words, we supposed that AB would "slide" from keratins to frog proteins. The result of this experiment is shown in Fig. 3b. Affinity-purified AB on keratins reveal a protein of 70 kD among the NE proteins; the same occurs for liver proteins and no signal is noted in the inner part of the oocyte nuclei. Thus, it has been shown in this experiment that MTBP has common antigenic determinants with keratins.Fig. 2. Vimentin, lamin, and MTBP in the frog oocyte nuclear preparations. 1, 2) Coomassie-stained 12% polyacrylamide gels after electrophoresis; Western-blot with antibodies against vimentin (3, 4), lamin (5, 6), and MTBP (7, 8); 1, 3, 5, 7) nuclear envelopes; 2, 4, 6, 8) oocyte nucleoplasm. On the left, molecular weights of standard proteins in kD.
This conclusion lead us to perform affinity purification of AB against MTBP on a column with keratins attached. This AB was used in the following work. Under the optimal dilution of 1:105, AB stained one protein in nuclear preparations from frog somatic blood and liver cells, with decreasing dilution to 0.8:105 two proteins (70 and 120 kD) were stained, and in dilution 1:104 cytokeratins were stained in addition to these two proteins (Fig. 3b).Fig. 3. Competitive binding (a) and testing of the affinity-purified antibodies (b). a: 1, 2) Keratins from human skin; 3, 7) nuclear envelopes; 4, 8) oocyte nucleoplasm; 5, 9, 6, 10) frog liver nuclei (two different isolations, respectively); 1-6) Coomassie-stained 10% polyacrylamide gels after electrophoresis; 7-10) Western-blot of the nuclear preparations after competitive binding (10% polyacrylamide gel). On the left, molecular weights of standard proteins in kD. b) Western-blot with the antibodies affinity-purified on human skin keratins. Frog liver nuclear preparation was loaded; 1) working AB dilution is 0.8:105; 2) AB dilution is 1:104.
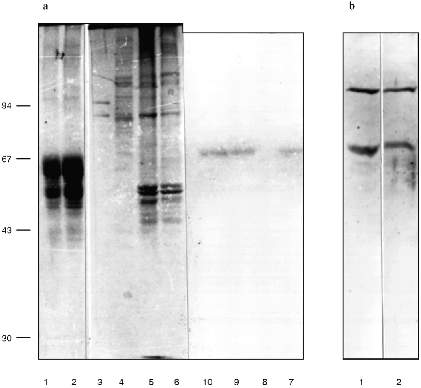
We conclude that the 70 kD protein which has common antigenic determinants with keratins is responsible for the telomere-binding activity in the NE extracts. We will call it membrane telomere-binding protein (MTBP).
MTBP Localization on the Oocyte Nuclear Envelope. NE spreads were prepared (see "Materials and Methods") for immunofluorescence experiments. It could be seen that AB stain the NE in a distinct pattern--a kind of network of connected circles made of double dots. The pattern is visible on well spread (not jammed) parts of the NE (Fig. 4a). There is a scheme of the staining in Fig. 4d.
Immunogold electron microscopy was done on ultrathin sections of the oocyte, while staining with AB against Xenopus lamins was used as a control. It can be seen in Fig. 5a that lamin is attached to the membrane bilayer from the inner part of the NE. The cytoplasmic outer part on the NE can be distinguished according to the position of yolk granules, the inner part according to the position of chromatin fibers. Oocyte NE is in abundance in comparison with the somatic nuclear membranes, and therefore the greater part of it is heavily folded [3]. Gold particles, which mark the position of MTBP, were never observed on the smooth parts on the NE. They are always connected with the electron-dense material protruding from the membrane to the cytoplasm. More often the signal is visible in membrane wrinkles, again from the NE cytoplasmic surface. It can be seen that the signal belongs to the tulip-like structures with a closed (Fig. 5b (1)) or an open (Fig. 5b (2)) cup. The scheme in Fig. 5c illustrates our understanding of the membrane organization according to the localization of antigens investigated.Fig. 4. Frog oocyte nuclear envelope: a) phase contrast; b) immunofluorescent staining; magnification ×400 in both cases; c) fragment of the NE stained with antibodies at higher magnification ×600; double dots are seen; d) schematic picture of staining.
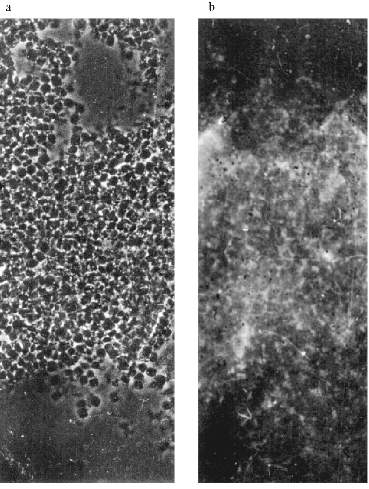
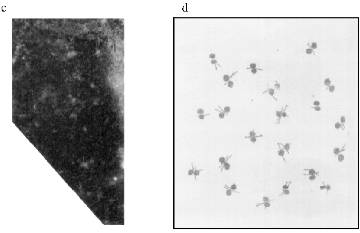
MTBP Localization in Somatic Cells. There is no permanent cell line of the frog Rana [22], so we used frog blood cells for immunofluorescence. Fully differentiated erythrocytes with heterochromatic nuclei are the main part of frog blood cells. It is possible to find among them lymphocytes with large vane-shaped nuclei and not fully differentiated cells of the erythropoietic gender [23]. Staining with AB against lamins was used as a control as before. The results of the experiment are shown in Fig. 6. In this case the pictures were taken with an Opton microscope. AB against lamins stain erythrocyte nuclei homogeneously; in the lymphocyte nuclei it is also possible to see lamin accumulations under nuclei membranes. Nonhomogeneous lamin staining has been observed in other vertebrate cells [19]. The pattern of AB against MTBP staining is fundamentally different. In this case the antigen is localized in nuclei and stains dots, often double-dots, or fields of dots that may be fused. Here we were not able to clarify if the staining belongs to the nuclear surface only. We could not determine the signal from the heterochromatic erythrocytes nuclei with the resolution available on the Opton microscope.Fig. 5. Immunogold staining of the frog oocyte: a) antibodies against lamin; magnification ×100,000; b) antibodies against MTBP; magnification ×60,000 (1) and ×50,000 (2); c) schematic diagram of the antigens localization; circles, lamin in the fibrillar material at the inner surface of the NE; asterisk, MTBP is located in the extended cup-like structures at the cytoplasmic surface of the NE.
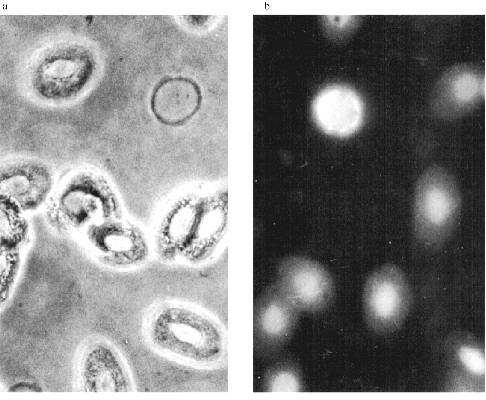
The Zeiss Axioplan microscope was used in subsequent work. The staining of the blood cells is shown in Fig. 7. DNA in the nuclei is stained with DAPI. MTBP is localized on the nuclei periphery, often in fused aggregates, though separate double dots can also be observed. The fused aggregates are built of double dots as is visible on the optical section through the nucleus.Fig. 6. Frog blood cells (Opton microscope): a) phase contrast; b) immunofluorescence with antibodies against lamin (magnification ×1000); c) immunofluorescence with antibodies against MTBP; 1) total preparation (×1000); 2) lymphocyte (×1500).
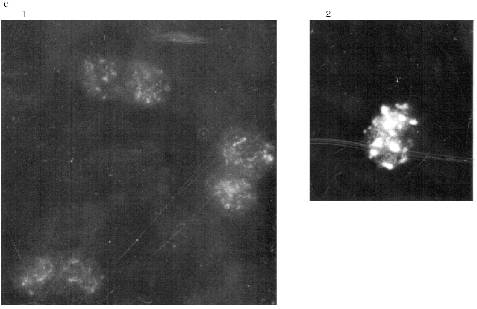
Double staining was done to determine the relative localization of the telomere sequences and MTBP. Green staining belongs to the secondary AB which reveal MTBP and is conjugated with FITC. The in situ hybridization was done with biotinylated vertebrate telomere fragment (T2AG3)135. In this case the staining was red because Texas Red-conjugated avidin was used to reveal the hybridization signal. The yellow color marks the overlapping signals. It is visible that the signals colocalize absolutely. Most of the telomeres occupy the periphery of the nucleus and are fused according to the position of telomere sequence as well as to the position of MTBP in the erythrocytes. The picture is more complicated in the lymphocyte nuclei. Telomeres and MTBP are also near the nucleus surface. They appear to be arranged in two round areas with a junction in between. Probably, there are several fused telomeres in each round area. More investigations should be done for a detailed localization of telomeres and MTBP in differentiated somatic cells, but there is no doubt in the colocalization of telomeres and MTBP in frog blood cell nuclei.Fig. 7. Immunofluorescence (a, b) and double labeling (c, d) of the frog blood cells (Zeiss Axioplan microscope). a) Optical section through lymphocyte nucleus, antibodies against MTBP (distinct double dots, fused areas of dots, and external localization of the antigen are visible); b) group of erythrocytes nuclei, anti-MTBP; c, d) double labeling (erythrocyte nuclei (c), lymphocyte nuclei (d)); 1) DAPI staining (blue); 2) anti-MTBP (green); 3) hybridization with (T2AG3)135 fragment (red); 4) combined image (yellow). Magnification ×2000. The colocalization of the anti-MTBP and telomere fragment is visible.


DISCUSSION
How We Found the "Right" Protein Using a "Wrong" Sequence. The first telomere cloned was from the ciliate Tetrahymena [24]. Very quickly it was found that the sequence (T2G4)n can function as a telomere in heterologous systems, for example, in yeast plasmids YAC [9, 25]. Then telomeres of higher eukaryotes were cloned and the high conservativeness of the sequence (T2AG3)n in the type of vertebrates was shown [26]. There has been no special investigation of frog telomeres, but it is reasonable to suppose that they consist of the same sequence. This has been shown for the toad Xenopus laevis [25].
The cross-hybridization between (T2G4)n sequence from Tetrahymena telomere and (T2AG3)n sequence from human telomere has also been demonstrated [27]. The same article states that part of the hybridization signal possesses Bal-sensitivity, i.e., belongs to the telomere, and the other part does not, i.e., it belongs to the intrachromosome areas with high homology to the (T2G4)n sequence. One of the pictures of this article represents hybridization of the restricted genomic DNA from different species with a labeled (T2G4)130 fragment. Most of the species, murine species for example, possess intrachromosomal sequences with high homology to (T2G4)130, but surprisingly there are no such sequences in the toad Xenopus laevis DNA and therefore the whole signal reflects hybridization between (T2G4)130 and (T2AG3)n [27]. Probably, the same is correct for the frog Rana temporaria. That is why the protein we picked up happens to posses specificity to the vertebrate telomere sequence (T2AG3)n in spite of the probe (T2G4)130 used at the beginning of the work. This is indicated, for example, by the experiments with double labeling.
MTBP and Keratins Share Common Antigenic Determinants. The intermediate filament type proteins including lamins have DNA-binding activity [19]. Attempts to find out if this activity is specific towards any exact sequences are going on constantly. Lamins have some selectivity towards oligonucleotides which mimic telomeric sequences [28]. Some evidence such as extraction properties and the picture of raw serum staining suggested that we were dealing with a protein that has something in common with intermediate filament type proteins. It has been found that there are no common antigenic determinants between MTBP with lamin or vimentin (Fig. 2), but serum does react with human skin keratins. This fact was used for affinity purification of AB (Fig. 3).
Keratins exist as heterodimers and it is known that there is a special gene for each chain. Characteristic sequences termed "head", "rod", and "tail" are distinguished inside these genes. In the article cited [29], a keratin-depleted cell line was transformed with a number of constructs containing keratin genes with sequential deletion from "tails" and "heads". Keratin localization in stable transformants was determined with the help of antibodies against human cytokeratins which genes were used for construct production.
The authors of [29] made a very interesting observation. The protein products of the control undeleted genes form filaments and in immunofluorescence give the normal picture of cytoplasmic localization but, along with the shortening of the gene, the staining becomes both intranuclear and cytoplasmic at first and then only intranuclear. The pictures of intranuclear staining are different for constructs with different deletions [29]. There are pictures among those given in the article which are surprisingly reminiscent of staining on MTBP and the well-known telomere-binding protein TRF1 [30]. The authors discuss the possibility that there may exist soluble or globular proteins unable to form conventional filaments, without a nuclear localization signal, but which share common antigenic determinants with cytokeratins and have a nuclear localization. The authors wonder if there are natural analogs of these artificially constructed proteins [29]. The results of the current work give an answer to that question--at least one of such proteins, MTBP, exists.
Comparison of MTBP with Tbf1 and TRF1. In our experiments antibodies (AB) obtained against MTBP from frog oocyte NE happen to be not species-specific. We identified a telomere-binding protein in extracts from mouse nuclear matrix using the gel shift assay, Western-blot with AB against MTBP, and column chromatography [31]. The identity of this protein with the earlier described telomere-binding protein Tbf1 was proved with the AB kindly provided by Dr. E. Gilson. Tbf1 was originally isolated from yeast but it exists in all the species investigated [5, 31]. The telomere-binding protein TRF1 [30] could be the same protein as MTBP and Tbf1 despite of the difference in the molecular masses published. It is possible also that there are different proteins of the telomere-binding complex which share the same "telobox" motif. Detailed comparison of the proteins isolated in different laboratories is still needed.
A very brief summary of the existing information about telomere-binding protein(s) is as follows. In preparations from mouse and human cells the protein(s) exist as a double line with approximate molecular mass 65 kD [5] or 70 kD according to our data [31]. Genes for the proteins are cloned and it is found that special DNA motif named "telobox" is responsible for binding to the telomere; the telobox has some features similar to the DNA-binding part of the proteins from the Myb-family, the family of transcriptional factors which play a role in oncogenesis [32], but the telobox is unable to bind Myb-specific oligonucleotides [5]. Antibodies against TRF1 stain dots and often double dots in the interphase nuclei of the cell culture, which correspond to the positions of the telomeres according to in situ hybridization. TRF1 is shown to be localized at the ends of metaphase chromosomes from the transformed cells with the construct containing the TRF1 cloned gene [30].
The results of the current work add to the protein(s) characteristics, that it is situated on the oocyte nuclear membrane and, probably, on the nuclear membrane of somatic cells. The fact that nuclear membrane localization has not been proved yet in cells in culture [33] may be attributed to the model used, i.e., all the cells in culture are malignant, and some other details of the method. MTBP, which is identical to Tbf1 according to AB cross-reactivity, is a membrane protein in the frog oocytes nuclei and is an integral part of the mouse nuclear matrix; at the metaphase plate of the nontransformed cell line MTBP exists on patches of the residual membrane structure; MTBP is revealed on the surface of the isolated mouse liver nuclei [31].
The century old story of the Rabl model is far from being completely understood. It is known now that there are cells with rigidly fixed chromatin organization such as spermatozoids [34, 35] or embryonic cells [36]; at the same time there are cell types which show less precise chromatin organization [37]. Nevertheless, in cases when telomeres are attached to the nuclear membrane, i.e., in oogenesis, MTBP (or Tbf1) is the main part of the protein complex which is responsible for that attachment.
We are very grateful to the staff members of the Institute of Cytology (Russian Academy of Sciences) A. P. Voronin and I. B. Lobov for the help in methods and to Dr. D. B. Gromov for a fruitful discussion. We would like to thank also Dr. A. R. Mitchell (HGU MRC, Western General Hospital, Edinburgh) for the possibility to use equipment and software for double labeling and Dr. E. Gilson (Ecole Normale Superieure de Lyon, France) for the antibodies against synthetic oligopeptide, part of Tbf1.
This study was supported with grants from the Wellcome Trust (UK) and the DOE Human Genome Program (USA) and also with grants from the Russian Foundation for Basic Research.
LITERATURE CITED
1.Rabl, C. (1885) Morphlog. Jahrbuch, 10,
214-330.
2.Dernburg, A. F., Sedat, J. W., Cande, W. Z., and
Bass, H. W. (1995) Cytology of Telomeres. Telomeres, Cold Spring Harbor
Press, pp. 295-338.
3.Gruzova, M. N., Tsvetkov, A. C., Pochukalina, G.
N., and Parfenov, V. N. (1995) Tsitologiya, 37, 744-769.
4.Dumont, J. N. (1972) J. Morphol., 136,
136-164.
5.Belgrader, Ph., Siegel, A., and Berezney, R. (1991)
J. Cell Sci., 98, 281-291.
6.Bilaud, Th., Koering, C. E., Binet-Brasselet, E.,
Ancelin, K., Pollice, A., Gasser, S., and Gilson, E. (1996) Nucleic
Acids Res., 27, 1294-1303.
7.Krohne, G., Franke, W. W., and Scheer, U. (1978)
Exp. Cell Res., 116, 85-102.
8.Bradford, M. (1976) Anal. Biochem., 72,
248-254.
9.Murray, A. W., Schultes, N. P., and Szostak, J. W.
(1986) Cell, 45, 529-536.
10.Maniatis, T., Frich, E., and Sambrook, J.
(1984) Molecular Cloning [Russian translation), Mir,
Moscow, pp. 197-198.
11.Strauss, F., and Varshavsky, A. (1984)
Cell, 37, 889-901.
12.Laemmli, U. K. (1970) Nature, 227,
680-682.
13.Callan, U. G. (1986) Lumpbrush Chromosome,
Springer-Verlag, Berlin.
14.Macgregor, H. C., and Varley, J. M. (1986)
Working with Animal Chromosomes [Russian translation],
Mir, Moscow, pp. 146-148.
15.Kipling, D., Mitchell, A. R., Masumoto, H.,
Wilson, H. E., Nicol, L., and Cooke, H. J. (1995) Mol. Cell
Biol., 15, 4009-4020.
16.Mitchell, A. R., Jeppesen, P., Hanratty, D., and
Gosden, J. (1992) Chromosoma, 101, 333-341.
17.Gooderham, K., and Jeppesen, P. (1983) Exp.
Cell Res., 144, 1-14.
18.Bugaeva, E. A., Parfenov, V. N., and Podgornaya,
O. I. (1992) Mol. Biol. (Moscow), 26, 983-992.
19.Moir, R. D., Spann, T. P,. and Goldman, R. D.
(1995) Int. Rev. Cytol., 162B, 141-173.
20.Cress, A., and Kurath, K. (1988) J. Biol.
Chem., 263, 19678-19683.
21.Georgatos, S. D., and Blobel, G. ( 1987) J.
Cell. Biol., 105, 117-125.
22.Hay, R., Capito, J., Chen, T. K., Mary, M.,
McClintock, P., and Reid, Y. (eds.) (1992) American Type Culture
Collection: Cell Lines and Hybridomas, 7-th ed., Rockville,
Maryland.
23.Sakuta, G. A., and Goryshina, E. N. (1982)
Tsitologiya, 24, 1298-1304.
24.Greider, C. W., and Blackburn, E. H. (1987)
Cell, 51, 887-898.
25.Weber, S., Schmid, M., Meyer, J., Cooke, H. J.,
and Lipps, H. J. (1993) Cell Biol. Int., 17, 623-624.
26.Kipling, D. (1996) The Telomere, Oxford
University Press, Oxford.
27.Allshire, R. C., Gosden, J. R., Cross, S. H.,
Granston, G., Rout, D., Sugawara, N., Szostak, J. W., Fantes, P. A.,
and Hastie N. D. (1988) Nature, 332, 656-659.
28.Shoeman, R. L., and Traub, P. (1990) J. Biol.
Chem., 265, 9055-9061.
29.Bader, B. L., Magin, T. M., Freudenmann, M.,
Stumpp, S., and Franke, W. W. (1991) J. Cell Biol., 115,
1293-1307.
30.Chong, L., van Steensel, B., Broccoli, D.,
Erdjument-Bromage, H., Hanish, J., Tempst, P., and de Lange, T. (1995)
Science, 270, 1663-1667.
31.Podgornaya, O. I., Bugaeva, E. A., Lobov, I. B.,
Voronin, A. N., Gilson, E., and Mitchell, A. R. (1998) J. Cell
Biol., in press.
32.Smith, S., and de Lange, T. (1997) Trends
Genet., 13, 21-25.
33.Luderus, M. E., van Steensel, B., Chong, L.,
Sibon, O. C., Cremers, F. F., and de Lange, T. (1996) J. Cell
Biol., 135, 867-881.
34.Zalensky, A. O., Allen, M. J., Kobayashi, A.,
Zalenskaya, I. A., Balhorn, R., and Bradbury, E. M. (1995)
Chromosoma, 103, 577-590.
35.Zalensky, A. O., Breneman, J. W., Zalenskaya, I.
A., Brinkley, B. R., and Bradbury, E. M. (1993) Chromosoma, 102,
509-518.
36.Marshall, W. F., Denburg, A. T., Harman, B.,
Agard, D. D., and Sedat, J. W. (1996) Mol. Biol. Cell., 7,
825-842.
37.Manuelidis, L. (1990) Science, 250,
1533-1540.