Blockade of Telomerase Function by Nucleoside Analogs
Y. E. Yegorov,1,2 D. N. Chernov,1,3 S. S. Akimov,1 A. K. Akhmalisheva,4 Y. B. Smirnova,1 D. B. Shinkarev,1 I. V. Semenova,4 I. N. Yegorova,1 and A. V. Zelenin1
1Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, ul. Vavilova 32, Moscow, 117984 Russia; fax: (095) 135-1405; E-mail: yegorov@genome.eimb.rssi.ru2To whom correspondence should be addressed.
3Center of Medical Studies of Oslo University, ul. Vavilova 32, Moscow, 117984 Russia.
4Department of Embryology, School of Biology, Lomonosov Moscow State University, 119899 Russia.
Submitted June 30, 1997.
Two types of spontaneously transformed cells appear in the culture of senescent mouse embryonic fibroblasts. The first type are cells with restricted proliferative potential (up to 30 population doublings); the other type are immortalized cells. Cells of the first type, unlike those of the second, have no telomerase activity and undergo two rounds of senescence. Spontaneous transformation of mouse embryonic fibroblasts in the presence of the reverse transcriptase inhibitors azidothymidine and carbovir led to the formation of telomerase-free clones. A fraction of these clones have the ability to overcome senescence via the acquisition of high telomerase activity. Cells with a very high level of telomerase activity become resistant to azidothymidine and carbovir. Azidothymidine-induced artificial senescence of rat myoblasts in culture resembles the senescence of fibroblasts, but the resulting cells acquire sharp morphological peculiarities. The blockade of telomerase function by azidothymidine in human U-937 and MeWo cells leads to the shortening of telomeres, but does not result in senescence. A hypothesis of the generation of the signal that induces senescence is proposed. This hypothesis suggests a change in DNA conformation during telomere shortening as a result of a change of loop structure of telomeric chromatin.
KEY WORDS: telomerase, senescence, reverse transcriptase inhibitors, cell culture, differentiation, telomeres, spontaneous transformation.
Abbreviations: TAC) telomerase activity; RTI) reverse transcriptase inhibitors; MEF) mouse embryonic fibroblasts; PD) population doubling; AZT) 3´-azido-3´-deoxythymidine; ddC) 2´,3´-dideoxycytidine.
Telomeres of vertebrate chromosomes consist of hundreds to thousands of
tandem repeats of TTAGGG fragments [1]. It has been
suggested that their function is chromosome end protection and
prevention of chromosome fusion [2, 3]. Analysis of terminal restriction DNA fragments has
shown that the chromosomes of somatic cells lose from 50 to 200
nucleotides of TTAGGG sequence per cell division [4, 5]. These changes were
explained on the basis of the under-replication of the chromosome ends
due features of the molecular mechanism of DNA replication [6, 7]. The lagging chain of the
replicative fork cannot be synthesized through the 5´-end in the
presence of riboprimer, which, in turn, cannot be synthesized at is
very end. Losses of the end DNA make impossible unlimited
proliferation. The data suggest that the chromosome shortening to some
degree might induce cell senescence, and the telomere length is a
measure of proliferative potential [6, 8, 9].
In contrast to somatic cells with limited life-span, most immortal cells contain a special enzyme, telomerase (DNA-nucleotidyl exotransferase, EC 2.7.7.31) [10]. It is a ribonucleoprotein complex that synthesizes short repetitive sequences (in the case of vertebrates, TTAGGG) onto chromosomal 3´-ends [11-13].
The enzyme uses its own RNA template and hence is a kind of reverse transcriptase [14]. We proposed and proved that reverse transcriptase inhibitors (RTI)5 can block telomerase function and induce so-called artificial senescence [15-17]. However, it was not clear whether it was possible to determine similar data in other cells, particularly in human cells.
The present work deals with the further development of experiments with RTI on cells in different states of differentiation and of different species.
MATERIALS AND METHODS
Cells. Primary culture of mouse embryonic fibroblasts (MEF) was initiated from 17-19-day-old CBA mouse fetuses as described in [18]. 3T3 Swiss cells were obtained from ICRF (London), 3T3 NIH and U-937 cells from ATCC (USA). U-937 is a suspension culture of promyelocytic human cells capable of differentiation into macrophages [19]. MeWo are human tumorigenic melanoma cells, the gift of Dr. M. M. Moisenovich (Moscow State University). L-6 cells (rat skeletal myoblasts) were generously provided by Dr. I. I. Fridlyandskaya (Institute of Cytology, St. Petersburg). U-937 cells were grown in RPMI 1640 (Gibco, USA); other cells were grown in DMEM (Gibco). Both media were supplemented with 10% fetal calf serum (Gibco) and 40 U/ml of gentamicin. The cultures were grown at 37°C under an atmosphere containing 5% CO2.
Clones of spontaneously transformed cells were grown in tissue culture flasks (25 cm2) (Nunc, Denmark). The cells were replated using trypsin-EDTA when they achieved monolayer status. Microphotography was performed through a Diaphot (Nikon, Japan).
Telomerase Activity Determination. Telomerase activity (TAC) was measured as described in [17]. Cell extracts were prepared in as described in [12]. Extracts (10 µl) were mixed with reaction mixture (10 µl) containing 100 mM Tris-HCl (pH 8.2 at 4°C), 10 mM TTP, 10 mM dGTP, 2 mM MgCl2, 2 mM EGTA, 1 mM DTT, 2 mM spermine, 0.2 mM spermidine, 0.2 µM 5´-(TTAGGG)2-3´ primer, and 10 µCi [alpha-32P]dATP (4000 Ci/mmole). The samples were incubated at 30°C for 1 h. When the reaction had reached completion the mixture was transferred to DEAE paper (10 × 10 mm), washed with 5% disodium hydrogen phosphate, and dried. Radioactivity was determined in a scintillation counter. The results were standardized to the protein content in the extract as measured according to Bradford [20].
For RNA-independent activity determination, DTT was added to 5 mM, and extracts were treated with RNase A for 15 min at 37°C, 0.03 mg/ml. The value of TAC was found as the difference in radioactivity obtained in experiments with extracts not treated and pretreated with RNase A.
Proliferation rate measurement and the conditions for obtaining spontaneously transformed cells are described in [16, 17]. Telomere length was determination as described in [21].
RESULTS AND DISCUSSION
Experiments with Spontaneous Transformants. Rodent cells have a high ability to undergo spontaneous transformation [22, 23]. Resulting cells in contrast to the original cells usually possess telomerase activity [15]. To prove the influence of RTI on telomerase function, we investigated the process of spontaneous transformation in the presence of RTI. The results are published in part [16, 17].
Mouse embryonic fibroblasts (MEF) are cells with restricted proliferative potential. These cells senesce after ~20 population doublings (PD). Resulting non-dividing cells survive for a long time (months). After 2-3 months, with relatively high probability, compact areas of growth arise. The number of cells in these areas increases exponentially. These cells are spontaneously transformed MEF.
All clones of spontaneous transformants in our experiments can be divided into two groups: clones with restricted proliferative potential and immortal ones. Because of the restriction of the method we could not determine the exact quantity of short-living clones, but it is clear that most clones had proliferative potential below 20 PD. Nine clones with proliferative potential of 20-25 PD and three clones with proliferative potential of 25-30 PD were obtained. All the clones that overcame 30 PD level always grew well later; they all grew for more than 50 PD without changes in growth rate or morphology. In the initial stages of the investigation we cultivated five of such clones to the level of 100 PD and more; in subsequent experiments we froze clones at the level of 60-70 PD. So it was established that the proliferative potential of clones of spontaneous transformants changes discretely: it can be restricted to 30 PD or be unlimited (more than 100 PD).
Growth of clones with restricted proliferative potential slowly decreased a few passages before growth arrest; the cells enlarged in size and became very spread. Cell morphology and behavior resembled senescent cells. Large non-dividing cells survived for a long time. Their morphology changed in accordance with senescence by culture specific changes [24]. To take into account that spontaneous transformants arise from senescent cultures from long-time (2-3 months) non-dividing cells, it is possible to conclude that senescence in these cells occurs twice.
Measurements of telomerase activity (TAC) in cells of clones with restricted proliferative potential were done three times. All measurements showed the absence of TAC. TAC-free were one clone obtained in the presence of carbovir, one clone obtained in the presence of AZT, and one control clone. It is impossible with our method to measure TAC in the clones before 22-23 PD. To take into account that TAC-free cells should lose telomere sequences during proliferation, we suggest that the first and the second senescence take place in cells with different telomere length.
Figure 1 shows TAC of spontaneous transformants as a function of their age and conditions of culture. It is clear that spontaneous transformants obtained without RTI or in the presence of ddC (inactive to telomerase nucleoside [16]) have middle or increased TAC during the entire time of cultivation. There was only one exception; a TAC-free clone with restricted proliferative potential. However, the transformants obtained in the presence of RTI are telomerase-free at the initial steps of culture (up to 30 PD). Clones that initially grew in the presence of RTI and began to senesce after removal of RTI resumed their proliferation and acquired average TAC values [16, 17].
The results of our experiments suggest the existence of two ways for spontaneous transformation of MEF: with or without telomerase activation. In the absence of RTI the telomerase reactivation at the first stage is more likely for mouse cells in culture. In the presence of RTI the second way without initial telomerase reactivation becomes prevalent.Fig. 1. Dependence of telomerase activity of spontaneously transformed mouse cells on duration and condition of cultivation. Dark circles, spontaneously transformed cells cultivated in the presence of RTI; open circles, spontaneous transformants cultivated without RTI or in the presence of ddC. The lines connect the same clones.
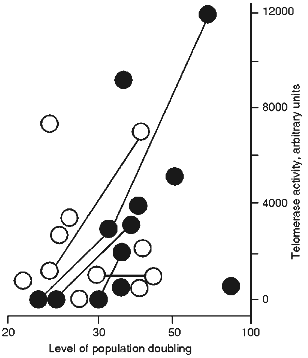
The telomeric hypothesis of senescence suggests the existence of feedback between telomere length and growth arrest [25]. The mechanisms of that feedback stop proliferation before complete physical disappearance of the telomeres [3, 26-28], and the weakening of these mechanisms can extend proliferative potential [26, 29, 30]. It is possible that in the case of telomerase-free clones an intracellular mechanism "estimating" telomeres change the scale; in this case short telomeres corresponding to the senescent state are accepted as sufficiently long.
The cases of very high TAC (TAC increased three times more than average) attract particular interest. Cells possessed such TAC level become resistant to RTI [16, 17] and usually appear after long cultivation.
The mechanism of RTI resistance due to elevating TAC suggests instability of the last telomeric nucleotide. That is the necessary condition for telomerase to lengthen telomeres in the presence of terminating nucleotides by multiple initiation processes. Protozoan and yeast telomerases possess 3´-exonuclease activity [31-33] and, besides, it is known that cycle-dependent telomere degradation can take place in telomerase-negative cells [34]. All of this can explain the last nucleotide instability and hence the appearance of RTI resistance.
One of the clones obtained had very low TAC, but it escaped AZT-induced senescence and grew in the presence of AZT for at least 70 PD. Such abnormal behavior of cells can be explained by the appearance of telomerase-independent AZT-resistance. Also, another explanation is possible: the cells of that clone may possess an alternative pathway of telomere maintenance [35].
During investigation of one of the clones, the following consequence of events happened. After two months of cultivation of this clone in the presence of AZT, the growth rate increased. This event coincided with the disappearance of TAC. TAC determinations were performed repeatedly after two weeks and one month with the same result. The culture became TAC-free. However, later, after ~30 PD from the acceleration of the growth, the proliferation of the cells gradually decreased. The culture began to resemble a senescent one. After removal of AZT the cells regained their proliferation and TAC appeared again. During the cultivation of cells of that clone without AZT, the cells proliferated stably for at least 100 PD and all that time they possessed TAC.
To explain the disappearance of TAC from the whole cell culture, we can propose that TAC-free cells have selective advantage in the presence of RTI, while their proliferation is not restricted by telomere length.
The main conclusion of interest from the experiments with spontaneous transformation is the following. Sometimes, in the course of spontaneous transformation, life-span extensions up to 30 PD occur.
Long-Term Cultivation of Various Non-fibroblastic Cells in the Presence of AZT. It was shown earlier that RTI can induce senescence-like processes in a culture of immortal mouse cells [16, 17]. 3T3 Swiss cells, NIH 3T3, and clones of spontaneously transformed MEF were studied. In most cases the sequence of events was similar: after a while proliferation began to decrease gradually; large, senescent-like cells appeared, their number increased. Finally, cell division ceased completely and for a long time (more than 4 weeks) the cells survived without division. It was interesting to know the result of such artificial senescence of non-fibroblastic cells.
For this purpose experiments with three types of cells were performed. We used cells of human promyelocytic leukemia U-937, human melanoma cells MeWo, and rat myoblast cells L-6.
The experiments with U-937 were performed in the presence of 1 and 2 µM AZT. The AZT concentration was determined according to [16, 17]. The proliferation decrease began after three weeks of cultivation. The ratio of attached cells increased and cell aggregates containing tens of cells appeared (Fig. 2, a and b). A considerable portion of cells had died, but proliferation continued. After approximately 5 months of cultivation the growth rate increased and the morphology of the culture became normal.
The experiments with MeWo cells were performed in the presence of 4 µM AZT. After one month of cultivation the proliferation slowly decreased and after two months growth rate sharply decreased; population size was constant for three weeks. There were proliferation and cell death simultaneously. Cell size changed insignificantly. Micronucleated cells appeared (Fig. 2, c and d). After a while the proliferation was restored.Fig. 2. Artificial senescence of U-937, MeWo, and L-6 cells under the influence of AZT: a) U-937, control; b) U-937, cultivated for two months in the presence of 2 µM AZT; c) MeWo, control; d) MeWo after two months cultivation in the presence of AZT; e) L-6 after two months of cultivation in the presence of AZT. The arrow shows a fibroblast-like cell. Phase-contrast; enlargement, ×370 (a-d) and ×180 (e).
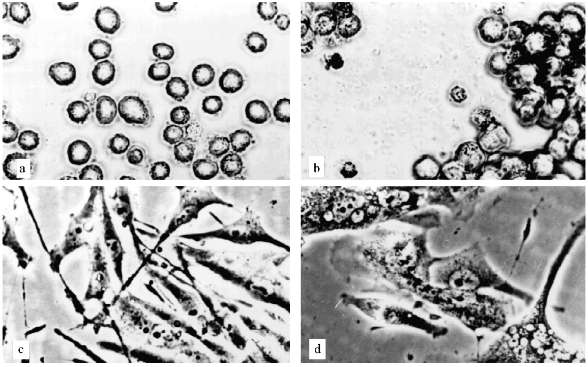
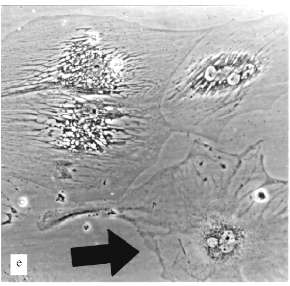
Thus, we could not reproduce the senescence phenomenon with human leukemia and melanoma cells. Even cells that stopped their proliferation, showed no significant alterations in morphology, no enlargement. Such non-dividing cells could not survive for a long time.
However, we revealed a long-term decrease of growth of human cells under the influence of AZT and demonstrated the telomere shortening after long AZT treatment [21]. The average telomere length in U-937 cells decreased in the presence of 1 µM AZT from 11 to 3 kb during 6 months. Average telomere length in MeWo cells decreased by ~1 kb after two months [21]. It is necessary to indicate that telomere length measurements were done at the time when cells restored their proliferation in the presence of AZT. To take into account that U-937 and MeWo cells were TAC-positive before and after the restoration of proliferation, it is probable that telomere length in time of long growth delay was even less, and that telomere shortening was the reason of the long growth delay.
It is known that complete telomere shortening can induce mechanisms of growth arrest that differ from ones that are induced during senescence, when telomere lengths are still several kilobases [36, 37].
Similar experiments on blocking telomerase function were also carried out earlier. Long exposure to AZT did not cause extreme shortening of telomeres and did not induce senescence in Tetrahymena cells [38]. Continuous expression of antisense RNA against the telomerase RNA component in HeLa cells resulted in cell death after ~25 population doublings (PD) [39]. Long-term cultivation of two immortal human lymphoid cell lines in the presence of different RTI led to telomere shortening but did not induced cell senescence [40].
It is more probable that the human cells used in these and our experiments lost the capability to senesce in the course of tumor progression. It is known that the most human tumor cells are immortal [10]. It was proposed that the immortalization process has two stages; during the first stage cells abandon or a neutralize senescence mechanism [37]. It is most probable that during telomere shortening in our experiments processes resembling crisis occurred. Cells are dying because of appearing genetic defects, but they continue to divide. The appearance of many micronucleated cells in MeWo culture indirectly indicates this possibility.
Thus, the blockade of telomerase function in human cells instead of senescence induction turn them to the previous stage of tumor progression that precede telomerase activation. These cells have altered growth regulation and are capable of proliferation with the telomere length corresponding to already non-dividing normal cells.
Epidemiologic data indicate that there are a few (on average 4) consequent events that happen in the course of carcinogenesis [41], and only one of these is telomerase activation. Taking into account the very hard control of telomerase repression in humans, it may be supposed with high probability that telomerase reactivation is not the first event in most cases of human carcinogenesis [42]. So telomere shortening in human tumor cells should not lead to the classical senescence when short telomeres induce the consequence of events that lead to non-division and specific alterations of morphology.
The experiments with L-6 cells were done in the presence of 5 µM AZT. After a few weeks of cultivation large cells appeared, the number of these cells increased, and they grew in size; the growth of the culture began to decrease gradually. At the end, after ~10 weeks, the culture mostly consisted of large non-dividing cells. The only difference between this culture and a culture of senescent fibroblasts was the morphology of the cells.
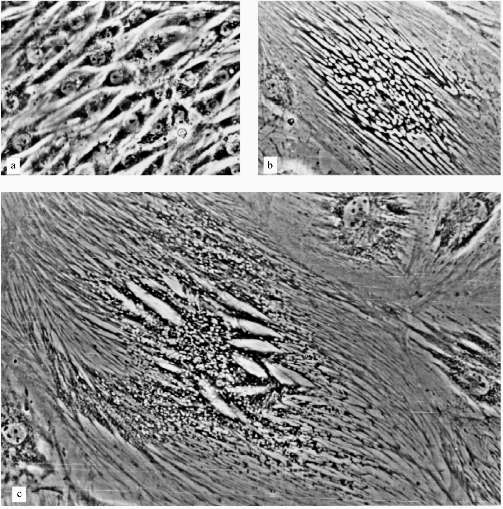
A very low number of large cells (less 1%) resembled senescent fibroblasts (Fig. 2e). Most of the large cells had very sharp morphological peculiarities. We observed viable cells with phase-contrast optics. From the early stages of the experiments increased development of cytoskeleton took place; there were concentration and polarization of cytoskeleton fibers. White, circle-shaped areas appeared in the cytoplasm (Fig. 3, b and c). Later, only in extremely large cells, highly organized structures resembling "plumes" appeared (Fig. 3c). Some of these cells were 0.8 mm long. Most of the cells were uninuclear. It took at least two weeks of non-division to obtain such giant cells.
The consideration of results of experiments with L-6 cells may be the following. There are two parallel processes in the cells: the process resemble senescence in fibroblasts and some myoblast-specific process. Strong development of cytoskeleton, the appearance of "plumes" can be considered as differentiation related processes, but happened in unusual conditions and so resulted in unusual cells. Probably, such differentiation-like processes can be induced in various cells by artificial senescence induction or in other ways, but this needs experimental confirmation.Fig. 3. Morphological changes of L-6 cells during artificial senescence: a) original culture; b, c) various non-dividing cells. Phase-contrast; enlargement ×370.
Differentiation-like processes can be induced in tumor cells by transition of the cells to non-division as a result of wild-type RB expression [43]. Cells of an osteosarcoma cell line with truncated RB gene after reexpression of transfected normal RB stopped their proliferation in early G1, enlarged in sizes, and became markedly spread. The expression of differentiation markers appeared. To take into account that the RB gene is involved in signal transduction pathways during senescence [25, 29], it can be assumed that there are common mechanisms involved in artificial senescence and after RB induction.
Hypothesis Concerning a Signal That Is Generated during Telomere Shortening. The main, unresolved question of telomeric theory of senescence is the nature of the signal that is generated in the course of telomere shortening.
It is known that mouse telomeres are very long (up to 100 kb and more) and hypervariable [44, 45]. This feature makes telomere analysis difficult. We proposed that during senescence the critical role belongs to the shortest telomeres. Then in the process of artificial senescence we can expect the appearance of such short (a few kilobases) telomeres comparable in size with these described for senescent human cells. The number of such short telomeres should be at least 1% of all telomeres, because there are about 100 telomeres in the investigated mouse cells.
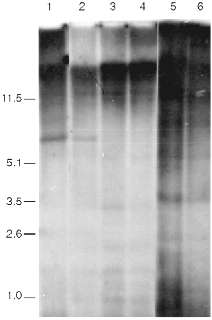
Data of analysis of telomeric repeats within terminal restriction fragments in senescence under the influence of AZT in 3T3 NIH cells, Swiss 3T3 cells, and cells of a clone of spontaneously transformed MEF are presented in Fig. 4. We cannot reveal any significant differences in the hybridization signal in the area 1-10 kb between the control and senescent cells. The high background level makes our goal (to measure with precision to within 0.5%) impossible. So, the question about the significance of ultralong mouse telomeres in the senescence remains unclear.
The main difficulty of the explanation of the nature of the signal that induces senescence arises from the fact that the telomere length of senescent mouse cells is too large and should (from the data obtained on human cells) correspond to the length of telomeres of young cells. Moreover, there are mouse lines with relatively short telomeres. Telomere dynamics in their cells correspond the human ones; during senescence in culture short telomeric fragments (a few kilobases) appear [46]. It is logical to suppose that there is shortening of ultralong telomeres during senescence of mouse cells, but due to the very large size the alteration of the average length should be too small (a few percent) to be measured by existing methods.Fig. 4. Southern blot hybridization of terminal restriction fragments during artificial senescence of mouse cells. Lanes: 1, 2) NIH 3T3; 3, 4) 3T3 Swiss cells; 5, 6) spontaneously transformed cells; 1, 3, 5) control cells; 2, 4, 6) cells after long-term cultivation in the presence of AZT. On the left, fragment sizes in kb.
The explanation of mouse telomeres behavior can be made by the existence of a supermolecular "gauge" of telomeres. A suitable candidate for this part is the nuclear matrix.
It is known that the TTAGGG sequence interacts with the nuclear matrix [47]. Recent studies showed that the human telomeric tracts bind to the nuclear matrix along their length and that the attachment sites occur frequently, on the order of one site per 1-2 kb [48]. The same mode of attachment was observed for mouse telomeres, but only within approximately half of their length. In the other half attachment sites are has rare [48]. Ultrastructurally, telomeres appear as compact nucleoprotein structures. Electron microscopy in situ hybridization to TTAGGG revealed "doughnut or horse shoe"-shaped telomere configurations ~100 nm long [48]. This probably indicates that telomeres form in space something like a cap. It was shown that in some cases (probably due to artifacts in preparations) telomere repeats can form very long linear tracts that even extend out of the nuclei [48].
There is a protein in the nuclear matrix that binds the telomeric repeat. It is TRF1 (TRF stands for TTAGGG Repeat Binding Factor) [48-50]. It was shown that in the case of hyperexpression of that protein, telomeric repeats gradually shorten; in the case of hyperexpression of defective in binding protein, telomeric repeats lengthen. Both effects take place despite constant TAC [51].
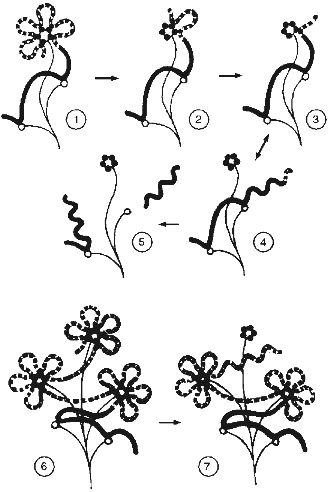
All these data help us to propose a model for telomere structure like the "camomile" (Fig. 5). The human telomeric repeat forms loops of about 2 kb that compactly localize near some nuclear matrix structure containing TRF. Similar loop size is determined by identity, monotony, and a feature of sequence (many GGG clusters) of telomeric repeats. In the course of telomere shortening, a special stage appears when telomeres become too short to form at least one loop. At this point senescence is induced [37]. Attachment of the telomeric sequence to matrix weakens, and the telomeric repeat can relatively easily dissociate to form a long free end.
It was suggested long ago that the intracellular signal that induces senescence can mimic the signal of DNA alteration [52]. DNA conformation in the free end of DNA should be similar with the conformation of DNA after a single-strand break. The length of the free end is determined by the next matrix attachment site, which can be rather remote. In this case the length of the free end should correspond to the usual size of DNA loops (hundreds of thousands of base pairs) [53]. The hypothesis predicts that a relatively short free DNA end with telomeric sequence is not recognized by the cell as an alteration region.Fig. 5. Scheme of relationships of telomeres and nuclear matrix (the "camomile" hypothesis). Bold lines, DNA; bold dotted lines, telomeric repeat; thin lines, nuclear matrix; dark circles, TRF; open circles, non-telomeric DNA--nuclear matrix attachment sites; twisted lines, altered DNA conformation; 1-5) human telomeres; 6, 7) mouse telomeres; 1, 6) states corresponding to young cells; 2) the last stable state during telomere shortening; 3, 4, 7) unstable conformations that induced senescence (M1); 5) state of crisis (M2).
Such long free ends should be generated predominantly at the end of mitosis, because the maximal loads on DNA--matrix connections should be at the end of chromosome decondensation. Assembly and disassembly of the loops take place in every cell cycle, so when the telomere length achieves 3 kb accidental restrictions of life-span appear. If such telomeres attach to the matrix by their middle part, the formation of loops become blocked.
The matrix attachment site of the long free DNA end should have more conformational freedom than the same site in the usual loop. Thus, as for usual matrix attachment sites, it should become the predominant target for endogenous nucleases [54]. As a result of nuclease attack and following under-replication, a matrix attachment site can be lost and a new long free DNA end can appear. Repetition of this process should lead to rapid DNA losses with speed corresponding to the loop size. This consequence of events can happen after unlimited telomere shortening during the crisis stage [37].
Probably, some features of organization of the loops of telomeric DNA reflect resistance of their matrix attachment sites to endogenous nucleases that is necessary for protection of all other internal loops.
To support the "camomile" hypothesis consider the following arguments.
1. The proposed model can explain the behavior of mouse telomeres. The three-dimensional structure of the telomere ("cap") has determined maximal size. In case of ultralong telomeres, telomeric sequence forms several "caps". In the course of telomere shortening in mouse the same events happen as in human cells, but the long free end appears when one "cap" is exhausted before the telomere sequence becomes very short. This is the reason that there are two types of matrix attachment sites in the mouse, i.e., with a distance ~2 kb apart and another type with longer distance.
2. According to the "camomile" hypothesis, senescence appears when the length of telomeres decreases to the extent when loop formation becomes impossible. Some telomeres without loop structure become the sources of signals that stop proliferation. In this case the alteration of the signal pathways can restore proliferation if the signal will be decreased or abandoned. However, life-span of the cell in this case will be restricted by the length of one telomeric loop. After complete telomere shortening, the free DNA end becomes the target of endogenous nucleases during the whole cell cycle. This will lead with high probability to cutting of DNA in the region of the matrix attachment site. DNA alterations become incompatible with life. This leads to crisis.
In the case of mouse cells, after increasing the life-span in accordance with the length of one loop, a long (few kilobases) free end of a telomeric sequence appears. This free end is protected against degradation (because it is telomeric), but has the wrong conformation that leads to repeated induction of senescence (M1), but not crisis (M2).
If we assume the average telomere loses about 50-100 bp per cell cycle, the length of one loop is sufficient for 20-40 divisions. This range (30 PD) was obtained in our experiments with spontaneous mouse transformants possessing restricted life-spans. Similar extensions of proliferative potentials of human cells were observed in cases of alterations of intracellular signal transductions due to expression of T-antigen SV-40, E1A and E1B antigens of adenovirus, papilloma virus antigens [30], or blocking pRb and p53 functions [25, 29].
3. The hypothesis can explain the accidental variation of proliferative potentials of single fibroblasts described by Smith et al. [55, 56]. During the last steps of senescence in culture of human fibroblasts many non-dividing cells appear. The process increases near the end. The accidental differences of proliferative potential between daughter cells appear when one of the daughter cells sharply stops divisions. This may be the result of wrong attachment of telomeres to the matrix, when in the presence of sufficiently long telomeres there is no loop formation.
4. Senescent cells stop their growth in the early G1 phase [57]. Disassembly of telomeric repeat and matrix should happen predominantly at the end of chromosome decondensation; this may lead to growth arrest at the beginning of G1.
5. According to the proposed hypothesis the generated during senescence signal is the same as after DNA alteration.
6. It is known that long telomeric DNA has nucleosomic structure that is damaged in the course of telomere shortening. The length of the mouse nucleosomic DNA within telomere is larger than the human ones [58]. It is clear that after alteration of the loop structure the nucleosomic structure should be altered too.
7. The hypothesis can explain the experiments with hypo- and hyperexpression of TRF1. After increasing the number of attachment sites the stabilization of telomere structure should happen with shorter telomeres and, in contrast, the telomeric repeat should be longer to form the loop structure with a decreased number of attachment sites.
8. Because the length of telomeric repeats in the cell and the quantity of TRF are controlled by different mechanisms, after formation of a hybrid cell the correspondence between total telomeric length and the quantity of attachment sites should be altered. This should destabilize DNA and can induce segregation of chromosomes with the shortest telomeres that usually was observed after somatic hybridization [59]. Previously we showed the segregation process in interphase [60].
The authors are grateful to S. M. Roberts for providing the carbovir, to N. F. Sullivan and to M. M. Moisenovich, and I. I. Fridlyandskaya for providing the cells. This work was supported by the Russian Foundation for Basic Research (grants No. 94-04-12843, 96-04-48277, and 96-04-49086) and scholarships from the Russian government.
LITERATURE CITED
1.Blackburn, E. H. (1991) Nature, 350,
569-573.
2.Zakian, V. A. (1989) Ann. Rev. Genet.,
23, 579-604.
3.Counter, C. M., Avilion, A. A., LeFeuvre, C.,
Stewart, N. G., Greider, C. W., Harley, C. B., and Bacchetti, S. (1992)
EMBO J., 11, 1921-1929.
4.Harley, C. B., Futcher, A. B., and Greider, C. W.
(1990) Nature, 345, 458-460.
5.Allsopp, R. C., Chang, E., Kashefi-Aazam, M.,
Rogaev, E. I., Piatyszek, M. A., Shay, J. W., and Harley, C. B. (1995)
Exp. Cell Res., 220, 194-200.
6.Olovnikov, A. M. (1971) Dokl. Akad. Nauk
SSSR, 201, 1496-1499.
7.Watson, J. D. (1972) Nature, 239,
197-201.
8.Allsopp, R. C., Vaziri, H., Patterson, C.,
Goldstein, S., Younglai, E. V., Futcher, A. B., Greider, C. W., and
Harley, C. B. (1992) Proc. Natl. Acad. Sci. USA, 89,
10114-10118.
9.Harley, C. B. (1991) Mutat. Res.,
256, 271-282.
10.Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., Ho, P. L. C., Coviello, G. M., Wright, W.
E., Weinrich, S. L., and Shay, J. W. (1994) Science,
266, 2011-2014.
11.Greider, C. W., and Blackburn, E. (1985)
Cell, 43, 405-413.
12.Morin, G. B. (1989) Cell, 59,
521-529.
13.Meyne, J., Ratliff, R. L., and Moyzis, R. K.
(1989) Proc. Natl. Acad. Sci. USA, 86, 7049-7053.
14.Greider, C. W., and Blackburn, E. H. (1989)
Nature, 337, 331-337.
15.Chernov, D. N., Yegorov, Y. E., and Akimov, S. S.
(1996) Dokl. Ros. Akad. Nauk, 349, 121-123.
16.Yegorov, Y. E., Chernov, D. N., Akimov, S. S.,
Bolsheva, N. L., Krayevsky, A. A., and Zelenin, A. V. (1996) FEBS
Lett., 389, 115-118.
17.Yegorov, Y. E., Chernov, D. N., Akimov, S. S.,
Bolsheva, N. L., Kraevsky, A. A., and Zelenin, A. V. (1997) Mol.
Biol. (Moscow), 31, 130-136.
18.Prudovsky, I. A., Yegorov, Y. E., and Zelenin, A.
V. (1985) Cell Differentiation, 17, 239-246.
19.Sundstrom, C., and Nilsson, K. (1976) Int. J.
Cancer, 17, 565-577.
20.Bradford, M. M. (1976) Analyt. Biochem.,
72, 248-254.
21.Yegorov, Y. E., Akhmalisheva, A. K., Smirnova, Y.
B., Shinkarev, D. B., Chernov, D. N., Zelenin, A. V., and Kraevsky, A.
A. (1997) Genetika, 33, 1433-1435.
22.Kuroki, T., and Huh, N. (1993) Jap. J. Canc.
Res., 84, 1091-1100.
23.Curatolo, L., Erba, E., and Morasca, L. (1984)
In vitro, 20, 597-601.
24.Bayreuher, K., Rodemann, H. P., Hommel, R.,
Dittmann, K., Albiez, M., and Francz, P. I. (1988) Proc. Natl. Acad.
Sci. USA, 85, 5112-5116.
25.Shay, J. W., Pereira-Smith, O. M., and Wright, W.
E. (1991) Exp. Cell Res., 196, 33-39.
26.Shay, J. W., Wright, W. E., Brasiskyte, D., and
van der Haegen, B. A. (1993) Oncogene, 8, 1407-1413.
27.Levy, M. Z., Allsopp, R. C., Futcher, A. B.,
Greider, C. W., and Harley, C. B. (1992) J. Mol. Biol.,
225, 951-960.
28.Shay, J. W., van der Haegen, B. A., Ying, Y., and
Wright, W. E. (1993) Exp. Cell Res., 209, 45-52.
29.Hara, E., Tsurui, H., Shinozaki, A., Nakada, S.,
and Oda, K. (1991) Biochem. Biophys. Res. Commun., 179,
528-534.
30.Wright, W. E., and Shay, J. W. (1992) Exp.
Gerontol., 27, 383-389.
31.Collins, K., and Greider, C. W. (1993) Genes
Dev., 7, 1364.
32.Bednenko, J., Melek, M., Greene, E. C., and
Shippen, D. E. (1997) EMBO J., 16, 2507-2518.
33.Cohn, M., and Blackburn, E. H. (1995)
Science, 269, 396-400.
34.Wellinger, R. J., Ethier, K., Labrecque, P., and
Zakian, V. A. (1996) Cell, 85, 423-433.
35.Bryan, T. M., Englezou, A., Gupta, J., Bacchetti,
S., and Reddel, R. R. (1995) EMBO J., 14, 4240-4248.
36.Wright, W. E., Pereira-Smith, O. M., and Shay, J.
W. (1989) Mol. Cell. Biol., 9, 3068-3092.
37.Shay, J. W., Wright, W. E., and Werbin, H. (1993)
Breast Cancer Res. Treatment, 25, 83-94.
38.Strahl, C., and Blackburn, E. H. (1994)
Nucleic Acids Res., 22, 893-900.
39.Feng, J., Funk, W. D., Wang, S.-S., Weinrich, S.
L., Avilion, A. A., Chiu, C.-P., Adams, R. R., Chang, E., Allsopp, R.
C., Yu, J., Le, S., West, M. D., Harley, C. B., Andrews, W. H.,
Greider, C. W., and Villeponteau, B. (1995) Science,
269, 1236-1241.
40.Strahl, C., and Blackburn, E. H. (1996) Mol.
Cell. Biol., 16, 53-65.
41.Alberts, B., Bray, D., Lewis, J., Raff, M.,
Roberts, K., and Watson, J. D. (1994) Molecular Biology of the
Cell [Russian translation], Vol. 3, Mir, Moscow, p. 452.
42.Yegorov, Y. E. (1997) Mol. Biol. (Moscow),
31, 16-24.
43.Ookawa, K., Tsuchida, S., Adachi, J., and Yokota,
J. (1997) Oncogene, 14, 1389-1396.
44.Kipling, D., and Cooke, H. J. (1990)
Nature, 347, 400-402.
45.Starling, J. A., Maule, J., Hastie, N. D., and
Allshire, R. C. (1990) Nucleic Acids Res., 18,
6881-6888.
46.Prowse, K. R., and Greider, C. W. (1995) Proc.
Natl. Acad. Sci. USA, 92, 4818-4822.
47.De Lange, T. (1992) EMBO J., 11,
717-724.
48.Luderus, M. E. E., van Steensel, B., Chong, L.,
Sibon, O. C. M., Cremers, F. F. M., and de Lange, T. (1996) J. Cell
Biol., 135, 867-881.
49.Zhong, Z. L., Shiue, S., Kaplan, S., and de
Lange, T. (1992) Mol. Cell. Biol., 13, 4834-4843.
50.Chong, L., van Steensel, B., Broccoli, D.,
Erdjument-Bromage, H., Hanish, J., Tempst, P., and de Lange, T. (1995)
Science, 270, 1663-1667.
51.Van Steensel, B., and de Lange, T. (1997)
Nature, 385, 740-743.
52.Wright, W. E., and Shay, J. W. (1996) in
Cellular Aging and Cell Death, Wiley-Liss, Inc., pp.
153-166.
53.Cook, P. R., and Brazell, I. A. (1976) J.
Cell. Sci., 22, 287-302.
54.Gromova, I. I., Nielsen, O. F., and Razin, S. V.
(1995) J. Biol. Chem., 270, 18685-18690.
55.Smith, J. R., and Whitney, R. G. (1980)
Science, 207, 82-84.
56.Jones, R. B., Whitney, R. G., and Smith, J. R.
(1985) Mech. Ageing Dev., 29, 143-149.
57.Sherr, C. J., and Roberts, J. M. (1995) Genes
Dev., 9, 1149-1163.
58.De Lange, T. (1996) Seminars Cell Dev.
Biol., 7, 23-29.
59.Weiss, M., and Green, H. (1967) Proc. Natl.
Acad. Sci. USA, 58, 1104-1111.