Immortalized Cells with No Detectable Telomerase Activity. A Review
R. R. Reddel,1,2 T. M. Bryan,1,3 and J. P. Murnane4
1Children's Medical Research Institute, 214 Hawkesbury Rd., Westmead, Sydney, NSW 2145, Australia; fax: +61-2-9687-2120; E-mail: rreddel@mail.usyd.edu.au2To whom correspondence should be addressed.
3Current address: Department of Chemistry and Biochemistry, HHMI, University of Colorado, Boulder, CO 80309-0215, USA.
4Radiation Oncology Research Laboratory, 1855 Folsom St., University of California, San Francisco, CA 94103, USA; fax: +1 (415) 476-9069; E-mail: murnane@rorl.ucsf.edu
Submitted July 28, 1997.
Immortalization of human cells in culture is usually associated with expression of telomerase activity. In some cases, however, no telomerase activity is detectable even though comparison of the terminal restriction fragment (TRF) pattern before and after immortalization shows that lengthening of telomeres has occurred. The extreme heterogeneity in telomere length and the differences in the dynamics of telomere maintenance in telomerase-negative cell lines compared to telomerase-positive cell lines indicate that these cells have utilized one or more alternative mechanisms for lengthening of telomeres (ALT). All telomerase-negative immortalized cell lines examined to date show evidence of ALT activity, consistent with the hypothesis that telomere maintenance either by telomerase or by ALT is required for immortalization. The nature of the ALT mechanism(s) is currently unknown, but studies of telomere dynamics in an ALT cell line containing a marker just proximal to the telomeric sequences show gradual shortening of the telomere followed by rapid elongation. This is consistent with a non-reciprocal recombinational mechanism similar to that found in telomerase-defective mutant yeast strains.
KEY WORDS: telomeres, alternative lengthening of telomeres, ALT, telomerase, senescence, crisis, terminal proliferation arrest.
According to the telomere hypothesis of senescence, a permanent cell cycle arrest is triggered when one or more telomeres are shortened beyond a critical length [1-3]. Consistent with this hypothesis, there is experimental evidence that telomeres shorten when normal cells undergo replication [4]. The reasons for this telomere shortening are not entirely clear, but may include the so-called "end-replication problem" where removal of the most distal RNA primer is thought to result in failure of the last few bases of the telomere to be replicated [5], and the activity of a putative exonuclease that removes the terminal 100-200 nucleotides of the C-rich strand of telomeric DNA [6].
Whether the telomeres of senescent cells have truly shortened to a critical length is unclear, however, because experiments in which senescence is over-ridden by expression of DNA tumor virus oncoproteins [7-9] or by loss of wild-type p53 [10] have shown that telomeres can continue to shorten when further cell division occurs. Although the loss of function of genes involved in triggering senescence [11] can result in a temporary escape from senescence, after further proliferation and telomere shortening the cells enter another terminal proliferation arrest state known as crisis [12]. The increase in chromosome fusions and cell death associated with crisis suggest that the shortened telomere length of cells in crisis is incompatible with normal function.
Cells that escape from crisis are usually capable of unlimited proliferation, i.e., they are immortalized. A prediction of the telomere hypothesis of senescence is that immortalized cells must have some process for preventing further telomere shortening. In most immortalized cell lines, this process involves the activity of a multi-subunit enzyme, telomerase [13]. Telomerase uses an RNA template for synthesizing telomeric DNA repeats to replace those that are lost as a result of cell division. In some cell lines, however, no telomerase activity is detectable. In this article the evidence that these cell lines have an alternative mechanism for the lengthening of telomeres (ALT) will be reviewed.
In vitro Immortalized Cell Lines without Telomerase Activity
Some human cell lines immortalized in vitro were found to have no detectable telomerase activity (Fig. 1) [10, 14-17] in the sensitive telomeric repeat amplification protocol (TRAP) assay [15]. Mixing experiments yielded no evidence that these cell lines contain inhibitors of telomerase itself or of the TRAP assay [16]. Similarly, dilution of TRAP assay samples did not reveal evidence of the presence of telomerase inhibitors (Fig. 2).
Fig. 1. Telomerase activity before and after immortalization. Lysates from the human cell lines indicated were subjected to TRAP assay before crisis (pre) and after escape from crisis (post). The cell lines were derived from fibroblasts (BFT-3K, BFT-3I, IIICF-T/A6, and BFT-3B), mesothelial cells (MeT-5A), and bronchial epithelial cells (HB56B/5T, BET-3M, BES-1A1, and BET-1A). LB is the lysis buffer negative control; positive controls are lysates equivalent to 100 and 1000 HeLa cells. Telomerase activity is detected as a 6 base-pair ladder following electrophoresis of radiolabeled reaction products on a polyacrylamide gel. For each of the cell lines shown, no telomerase was detected before immortalization. In the majority, telomerase was seen after immortalization, but in some cells (IIICF-T/A6 and BET-3M) no telomerase activity was detectable.
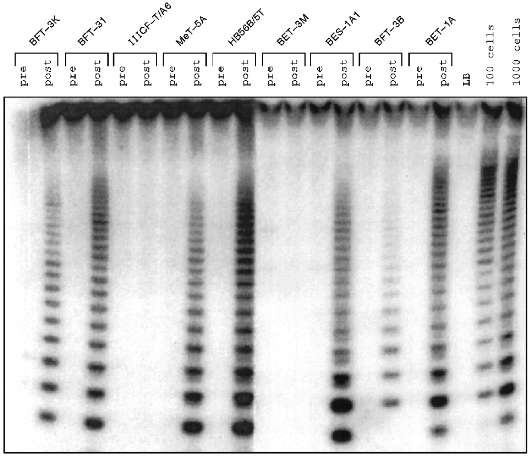
Although most of the cell lines studied are SV40-immortalized fibroblasts, lines derived from other cell types and immortalized in other ways (including the use of other DNA tumor virus genes, chemical carcinogens, and spontaneously in the case of fibroblasts from a Li--Fraumeni syndrome (LFS) family member) can also be telomerase-negative [16]. Although the number of epithelial and mesothelial cell lines studied so far is small, they seem more likely to be telomerase-positive than cell lines derived from fibroblasts [18].Fig. 2. Dilution of cell extracts did not reveal telomerase activity in a telomerase-negative immortalized cell line, IIICF/c. For cell lines IIICF/c and HeLa, the indicated amount of protein was assayed by TRAP. LB is the lysis buffer negative control, and lysates equivalent to 1000, 100, and 10 HeLa cells were used as the positive controls. Samples with 10 µg of HeLa extract had less TRAP activity than the lower concentrations indicating the presence of a dilutable inhibitor, but dilution of the IIICF/c extract did not reveal any evidence of telomerase activity.
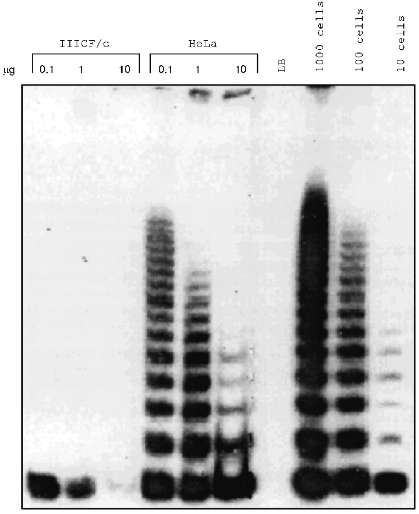
Overall, approximately one-quarter of all in vitro immortalized cell lines are telomerase-negative [18]. Some fibroblast strains, however, seem to have a propensity for immortalization in the absence of telomerase activity. Each of 10 cell lines derived from breast fibroblasts of an LFS individual by spontaneous immortalization or following transfection with SV40 or human papilloma virus HPV16 genes was telomerase-negative [16]. Eleven of 20 cell lines derived by SV40 gene transfection of jejunal fibroblasts from a non-LFS individual were telomerase-negative (P. Bonnefin et al., unpublished data). In one study 19 of 19 SV40-immortalized neonatal foreskin fibroblasts were telomerase-positive [19], but in another study 3 of 3 SV40-immortalized neonatal foreskin fibroblasts were telomerase-negative (J. Murnane, unpublished data). The reasons for these strain differences are not clear but could include p53 status (the LFS cells are heterozygous wild-type/null-mutant for p53) or the differentiation status of the cell strain.
Evidence for an Alternative Mechanism for Lengthening Telomeres (ALT)
Several lines of evidence indicate that the telomeres of telomerase-negative immortalized cell lines undergo lengthening. Telomere length is usually assessed either by gel electrophoresis or by chromosomal in situ hybridization. In the electrophoretic approach, genomic DNA digested with restriction enzymes that cut frequently throughout the genome but not within the telomeric repeat sequence is size-fractionated by electrophoresis and then probed with a labeled oligonucleotide (TTAGGG)n corresponding to multiples of the telomeric repeat sequence. This method detects terminal restriction fragments (TRFs) that consist of telomeric DNA plus a small amount of subtelomeric DNA between the telomere and the closest restriction site. Telomerase-negative immortalized cell lines have a TRF pattern that is distinct from immortalized cell lines with telomerase activity (Fig. 3): there is a smear from the bottom of the gel up to the well indicating that the TRF sizes range from very small to extremely large [16]. In situ hybridization studies using conventional [20] or peptide nucleic acid [21] probes have shown that this heterogeneity of telomere length occurs within individual telomerase-negative cells. The presence of the very long telomeres indicates that telomere lengthening has occurred. This characteristic pattern has been found in all telomerase-negative cell lines examined by TRF analysis to date and has never been seen in any telomerase-positive cell line where telomeres are generally much more homogeneous in length.
One possibility that has been considered is that the telomerase-negative cells maintain their telomeres by intermittent telomerase activity. If true, their telomeres would be expected to undergo shortening in the periods when telomerase is absent. However, no shortening was detected in one cell line, GM847, that was analyzed over the course of 98 population doublings (PD) [16]. In two other cell lines, IIICF-T/A6 and IIICF/c, there was evidence for an increase in the amount of large TRFs following immortalization and no subsequent decrease in mean TRF size despite the continued absence of detectable telomerase for 95 PD [16] and >600 PD ([10] and T. Bryan et al., unpublished data), respectively. Analysis of several additional telomerase-negative cultures before and after immortalization (Fig. 3) showed that in each case an increase in mean TRF size had occurred [16]. Since most of these cultures were clonally derived it is clear that telomere elongation had occurred, rather than the selection of a pre-existing clone with long telomeres.Fig. 3. Immortalized cell lines lacking telomerase activity have very long telomeres. Terminal restriction fragments (TRFs) were electrophoresed and detected with a radiolabeled telomere repeat sequence probe. Telomerase activity of the cell lines shown is indicated by "tel". The telomerase-negative immortalized cells all have very heterogeneous TRF lengths ranging from very short to very long (on the left, in kb). For two telomerase-negative cell lines, IIICF-T/A6 and BET-3M, TRF analysis is shown pre- and post-immortalization. In both cases, telomere lengthening has occurred following immortalization.
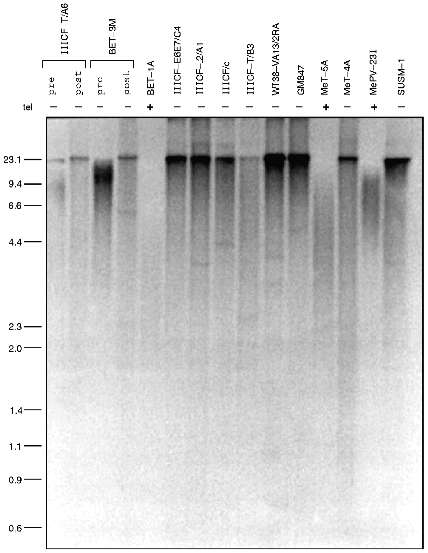
These data show that telomere elongation occurs in the absence of telomerase activity and thus indicate the existence of one or more alternative mechanisms for lengthening telomeres (ALT). Even though all of the ALT cell lines examined to date have a similar TRF pattern, it cannot be assumed at this stage that the mechanism of telomere lengthening is the same in each case.
In one telomerase-negative cell line, KB319, it has been possible to follow the dynamics of changes in the length of a single telomere [14]. This cell line has a plasmid inserted proximal to the repeat sequences of the telomere of chromosome 13q, so that probing of TRF gels with labeled plasmid detects the TRF pattern of only the marked chromosome. Three subclones of this cell line were studied: one with a long telomere on chromosome 13, one with a short telomere, and one in which no distinct band was discernible. These subclones were subcloned further and the TRF analyses showed the occurrence of both gradual and rapid length changes that sometimes involved many kilobases. The gradual changes were due to the shortening of telomeres at a rate consistent with that reported for telomeres of telomerase-negative normal somatic cells, eventually resulting in the loss of nearly all of the telomere. However, polymorphism was never detected in the subtelomeric plasmid sequences and no increase in the frequency of telomere associations occurred, indicating that the telomeres were not completely lost. Instead, the short telomeres were selectively elongated, indicating that a mechanism to recognize and salvage overly short telomeres exists in this cell line. The alternative possibility, that cells with complete telomere erosion were lost from the population seems unlikely because there was no indication of increased cell death or changes in cell doubling time during this process. The marked telomere also showed rapid, highly heterogeneous increases and decreases in length [14]. The frequency of these rapid changes in length varied in different subclones and showed a correlation with increased telomere associations, suggesting that the complete loss of telomeres could occur by these rapid events.
Despite different initial lengths, as a result of the dramatic increases and decreases in length, in different KB319 subclones the telomere marked by the plasmid insertion eventually took on the heterogeneous distribution in length characteristic of telomeres in cells utilizing ALT. The analysis of the dynamics of changes in the length of an individual telomere therefore demonstrates that telomerase-negative SV40-immortalized human cell lines continually maintain telomere length.
ALT in Tumor Cell Lines and Tumors
ALT is not restricted to in vitro immortalized cell lines, but has been found in 4 of 56 tumor-derived cell lines and 4 of 57 tumor samples (T. Bryan et al., unpublished data). Interestingly, 3 of the 4 ALT cell lines were derived from sarcomas. This is consistent with the observation that immortalized cell lines derived in vitro from fibroblasts are more commonly ALT-positive than lines derived from epithelial cells [18]. Since 90% of human tumors are of epithelial origin, this may account in part for the low incidence of ALT-positive tumors. There also appeared to be a possible association between ALT and tumors arising in Li--Fraumeni syndrome individuals (T. Bryan et al., unpublished data). The reasons for this putative association are not known since the majority of human tumors also contain p53 mutations but have telomerase activity. The first genetic "event" in the process of oncogenesis is the inherited p53 mutation in these cells, and maybe the order in which mutations occur is important in determining the probability of immortalization via the ALT pathway. The results of other studies are also consistent with the presence of ALT in tumors. For example, 2 of 56 renal cell carcinomas were telomerase-negative and contained cell clones with long TRFs [22].
A number of telomerase-negative tumors have been found to have telomeres that are much shorter than those characteristic of ALT (T. Bryan et al., unpublished data). It is therefore possible that some of these tumors do not contain immortalized cells or that there may be non-telomerase mechanisms of telomere maintenance that do not result in very long telomeres. Conversely, a minority of telomerase-positive tumors have very long telomeres indistinguishable from those of ALT. It is possible that mutations in a telomerase component or in a telomere-binding protein result in "runaway elongation" of telomeres by telomerase, as has been observed in Tetrahymena and the yeast Kluyveromyces lactis [23, 24]. Alternatively, there may be mixtures of ALT-positive and telomerase-positive cells in the same tumor, or ALT and telomerase may co-exist within the same cells. In the absence of direct assays for ALT, it is not presently possible to distinguish among these possibilities.
Possible Mechanisms of ALT
In considering the possible nature of the ALT mechanism(s), it may be instructive to note that in addition to telomerase there are two other mechanisms used by eukaryotes for maintenance of telomere length: recombination and retrotransposition. Wang and Zakian transferred telomere sequences from ciliates into the yeast, S. cerevisiae and demonstrated recombination of these sequences [25]. Consequently, they proposed that telomere--telomere recombination provides an efficient mechanism for rescue of DNA termini with very short stretches of telomeric DNA. S. cerevisiae strains lacking a functional EST1 gene underwent progressive telomere shortening and growth senescence, but a minor subpopulation was able to survive as the result of amplification and acquisition of subtelomeric elements [26]. This survival mechanism was shown to be dependent on RAD52, a protein involved in homologous recombination and double-strand break repair in a wide range of eukaryotes [27]. It was initially thought that this process might be restricted to S. cerevisiae because of the presence in this organism of large subtelomeric Y´ elements interspersed with telomeric repeats. However, deletion of the telomerase RNA gene from the yeast K. lactis, which does not contain subtelomeric telomere-like repeats also yielded survivors at a relatively high frequency. In this instance recombination of the telomere repeat sequences themselves was involved, which was also dependent on RAD52 [28]. The authors proposed that in the presence of telomerase activity the normal-length telomeres are capped with proteins that prevent recombination events, but that severely shortened telomeres become uncapped, promoting recombinational repair [28]. A similar mechanism was previously proposed to explain the rapid elongation of short telomeres in a telomerase-negative human cell line [14]. Similar mechanisms may be involved in telomere maintenance in telomerase-negative yeast and human cells, because the TRF pattern of the est1 S. cerevisiae and the telomerase-negative K. lactis resembled those seen in human ALT cell lines.
Analysis of the KB319 line suggests that ALT cells are capable of seeding new telomeres following chromosome breakage. Transfected plasmid DNA integrated at the end of a chromosome had telomeric repeat sequences added onto both ends, one end of which became the new telomere of this chromosome [29]. Other studies have also suggested that telomerase can mediate the de novo addition of telomere sequence in the healing of broken chromosomes [30]. In support of the possibility that ALT may involve recombination, it is of interest to note that addition of telomeres to broken yeast chromosomes may be mediated either by telomerase or by recombination [25, 31].
In Drosophila melanogaster and related dipterans non-LTR retroposons of the TART and HeT-A classes are utilized instead of short repeat sequences to replace the sequences lost from the end of chromosomes during cell division [32-37]. The telomeres of Drosophila are capped by chains of retroposons which undergo gradual shortening, and which, to compensate this loss, can be lengthened by a retrotransposition event. This mechanism is also able to heal broken chromosomes in Drosophila. Although it is not known whether retrotransposition can be utilized to lengthen telomeres in eukaryotes distant from Drosophila, an abnormally shortened telomere may well be perceived by the cell as a double-strand break and it has been shown that retroelements can mediate the repair of double-strand breaks in yeast in the absence of RAD52 [38, 39]. In yeast that are mutant at both the telomerase RNA gene and the RAD52 loci there are rare survivors which occur through a mechanism which is currently unknown [28]. Although it is possible that these survivors have retained some residual recombination activity, either RAD52-dependent or -independent, it will be interesting to determine whether retrotransposition is involved in maintenance of their telomeres.
The above data suggest that there are three telomere maintenance mechanisms (telomerase, retrotransposition, and recombination) that are conserved in most eukaryotes. Drosophila and related organisms might therefore be considered to be exceptions in which one of the three mechanisms, telomerase, has not been conserved and the retrotransposition mechanism which may serve as a "backup" in most other eukaryotes is preferentially used. However, the recent finding that the active site of yeast and Euplotes aediculatus telomerase is a reverse transcriptase that is closely related to the Drosophila TART retrotransposon, may suggest that the retrotransposition mechanism of telomere maintenance in Drosophila can be regarded as somewhat similar to the use of telomerase in other eukaryotes [40]. Thus, there may be only two main categories of telomere maintenance mechanisms--telomerase and recombination.
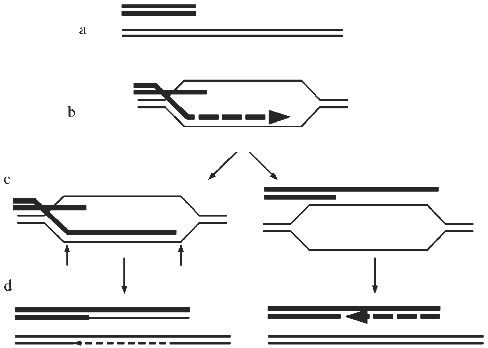
In view of the above considerations, both recombination and retrotransposition should be considered as candidate mechanisms of ALT. Detailed sequence data are not available for ALT telomeres at present, but hybridization of TRFs with telomere repeat probes has shown that they contain sequences that are closely related to the telomere repeat and that the amount of this sequence increases in ALT cells [16]. Therefore, recombination is more likely to be the mechanism of ALT than retrotransposition. A proposed model for recombination-mediated elongation of telomeres is shown in Fig. 4. It is important to keep in mind, however, that there may be more than one ALT mechanism.
A possibility that seems unlikely but cannot be ruled out at present is that ALT cells might contain an abnormal telomerase activity with the following properties: 1) its regulation is abnormal, resulting in telomere lengths ranging from short to very long; and 2) it is not able to be detected by conventional or TRAP assays. Evidence is accumulating for the role of telomere-binding proteins (TBPs) in feedback control of telomere length [41, 42], and certain mutations in the telomerase RNA gene (especially those that result in an abnormal telomere sequence that affects binding of TBPs) or mutations in TBPs themselves have been shown to cause "runaway elongation" of telomeres in Tetrahymena and the yeast K. lactis [23, 24, 43-46]. The combination of no detectable telomerase with heterogeneous and abnormally long telomeres could theoretically be produced if such mutations in a telomerase component or a TBP resulted both in dysregulated telomere elongation activity in vivo and loss of activity in the available in vitro assays. Alterations in TBPs were proposed as a possible mechanism for the rapid changes in the length of an individual telomere in a telomerase-negative cell line [14]. However, at present there is no evidence for changes in TBPs in cell lines expressing ALT, and indeed in some ALT cell lines there is no detectable expression of the human telomerase RNA (hTR) gene, making it very unlikely that telomerase is involved, at least in those cells [47]. Furthermore, in ALT cell lines that express hTR, no hTR mutations likely to cause dysregulated lengthening were found [47].Fig. 4. A model for recombination-mediated lengthening of telomeres [25, 28]. a) When a telomere becomes critically short it may be interpreted by the cell as a double-strand break (DSB). b) The DSB repair enzymes then mediate invasion by a single-stranded 3´ end of the short telomere between the strands of a longer telomere. This step may be dependent on RAD52. DNA polymerase may then extend the short strand, using the long strand as the template. c) The crossed-over strands may then be subject to cleavage by a nuclease (-->) followed by ligation, resulting in recombinant DNA molecules (d), as has been proposed for DSB repair by recombination in yeast [60]. Alternatively, the structure shown in (b) may be resolved by unwinding of the newly formed helix and rewinding (c), resulting in non-recombinant molecules (d). This has been shown to occur in DSB repair in yeast [61]. In either case, the staggered annealing of repeats in the short and long telomeres results in net telomere elongation.
Do Normal Cells Repress ALT?
Given the postulated importance of telomere maintenance to immortalization, and the well-documented ability of normal cells to suppress the immortal phenotype in normal--immortal hybrids, it seems likely that normal somatic cells contain repressors of ALT. The available data are consistent with this prediction, but definitive evidence is currently lacking. For example, a cell line GM847 that was subsequently found to be ALT-positive [16] exhibited a finite lifespan when fused with immortalized cell lines from other complementation groups [48-50], implying that telomere maintenance may have been repressed in the hybrids. When GM847 was fused with telomerase-positive immortalized cell lines, the senescent hybrid progeny were mostly telomerase-negative but there were insufficient cells to obtain DNA to determine whether the TRF pattern typical of ALT had also disappeared [16]. A large number of genes encoding helicases, topoisomerases and polymerases, have been identified that repress homologous recombination in yeast [51]. It is possible that the mammalian homologs of one or more of these genes may be responsible for repressing ALT in normal mammalian cells. In view of the apparent predilection for ALT in LFS cells, p53 must be considered as a potential contributor to the repression of ALT in normal cells. A possible mechanism may be suggested by the observation that both RAD52 and p53 bind RAD51 [52, 53].
We have addressed the question of whether the protein product of the ataxia-telangiectasia (AT) gene, ATM, might be involved in telomere maintenance. ATM has a relatively high degree of homology to the yeast TEL1 gene [54] which has been demonstrated to influence telomere length [55]. AT cells are reported to exhibit a high rate of end-to-end chromosomal fusions [56] and lymphocytes from AT individuals undergo accelerated telomere shortening [57], suggesting the possibility that ATM may normally be involved in telomere stability. Further, AT cells display elevated levels of intrachromosomal recombination [58]. Of 8 SV40-immortalized AT fibroblast lines, 5 were found to be telomerase-positive and 3 were ALT-positive indicating either that an SV40 gene product is capable of substituting for some aspect of ATM function or that ATM is not required for telomere maintenance via telomerase or ALT [59].
Clinical Implications of ALT
Regardless of the precise mechanism(s) involved, the existence of ALT has obvious implications for proposals to treat cancer with inhibitors of telomerase. Although ALT appears to be present in only a minority of tumors, effective inhibition of telomerase activity will subject tumors to strong selection pressure for the emergence of treatment-resistant cells via activation of ALT. A detailed understanding of ALT may make treatment with a combination of inhibitors of all telomere maintenance mechanisms feasible.
Work in the authors' laboratories was supported by the Carcinogenesis Fellowship of the New South Wales Cancer Council (to R. Reddel), a National Health and Medical Research Council of Australia postgraduate scholarship (to T. Bryan), and grant No. 3RO1CA69044-01S1 from the National Cancer Institute, NIH, USA. We thank Lindy Hodgkin for assistance in preparing the manuscript.
LITERATURE CITED
1. Olovnikov, A. M. (1971) Dokl. Akad. Nauk
SSSR, 201, 1496-1499.
2. Harley, C. B. (1991) Mutat. Res.,
256, 271-282.
3. Harley, C. B., Vaziri, H., Counter, C. M., and
Allsopp, R. C. (1992) Exp. Gerontol., 27, 375-382.
4. Harley, C. B., Futcher, A. B., and Greider, C. W.
(1990) Nature, 345, 458-460.
5. Levy, M. Z., Allsopp, R. C., Futcher, A. B.,
Greider, C. W., and Harley, C. B. (1992) J. Mol. Biol.,
225, 951-960.
6. Makarov, V. L., Hirose, Y., and Langmore, J. P.
(1997) Cell, 88, 657-666.
7. Counter, C. M., Avilion, A. A., LeFeuvre, C. E.,
Stewart, N. G., Greider, C. W., Harley, C. B., and Bacchetti, S. (1992)
EMBO J., 11, 1921-1929.
8. Counter, C. M., Botelho, F. M., Wang, P., Harley,
C. B., and Bacchetti, S. (1994) J. Virol., 68,
3410-3414.
9. Klingelhutz, A. J., Barber, S. A., Smith, P. P.,
Dyer, K., and McDougall, J. K. (1994) Mol. Cell. Biol.,
14, 961-969.
10. Rogan, E. M., Bryan, T. M., Hukku, B., Maclean,
K., Chang, A. C.-M., Moy, E. L., Englezou, A., Warneford, S. G.,
Dalla-Pozza, L., and Reddel, R. R. (1995) Mol. Cell. Biol.,
15, 4745-4753.
11.Duncan, E. L., and Reddel, R. R. (1997)
Biochemistry (Moscow), 62, 1477-1490 (Russ.).
12. Girardi, A. J., Jensen, F. C., and Koprowski, H.
(1965) J. Cell. Comp. Physiol., 65, 69-84.
13. Greider, C. W., and Blackburn, E. H. (1985)
Cell, 43, 405-413.
14. Murnane, J. P., Sabatier, L., Marder, B. A., and
Morgan, W. F. (1994) EMBO J., 13, 4953-4962.
15. Kim, N. W., Piatyszek, M. A., Prowse, K. R.,
Harley, C. B., West, M. D., Ho, P. L. C., Coviello, G. M., Wright, W.
E., Weinrich, S. L., and Shay, J. W. (1994) Science,
266, 2011-2015.
16. Bryan, T. M., Englezou, A., Gupta, J.,
Bacchetti, S., and Reddel, R. R. (1995) EMBO J., 14,
4240-4248.
17. Whitaker, N. J., Bryan, T. M., Bonnefin, P.,
Chang, A. C.-M., Musgrove, E. A., Braithwaite, A. W., and Reddel, R. R.
(1995) Oncogene, 11, 971-976.
18. Bryan, T. M., and Reddel, R. R. (1997) Eur.
J. Cancer, 33, 767-773.
19. Montalto, M. C., and Ray, F. A. (1996)
Carcinogenesis, 17, 2631-2634.
20. Henderson, S., Allsopp, R., Spector, D., Wang,
S.-S., and Harley, C. (1996) J. Cell Biol., 134,
1-12.
21. Lansdorp, P. M., Poon, S., Chavez, E.,
Dragowska, V., Zijlmans, M., Bryan, T., Reddel, R., Egholm, M.,
Bacchetti, S., and Martens, U. (1997) in Ciba Foundation
Symposium, No. 211, Telomeres and Telomerase (Cardew, G.,
ed.) Wiley-Liss, London, in press.
22. Mehle, C., Piatyszek, M. A., Ljungberg, B.,
Shay, J. W., and Roos, G. (1996) Oncogene, 13,
161-166.
23. Yu, G.-L., Bradley, J. D., Attardi, L. D., and
Blackburn, E. H. (1990) Nature, 344, 126-132.
24. McEachern, M. J., and Blackburn, E. H. (1995)
Nature, 376, 403-409.
25. Wang, S.-S., and Zakian, V. A. (1990)
Nature, 345, 456-458.
26. Lundblad, V., and Blackburn, E. H. (1993)
Cell, 73, 347-360.
27. Petes, T. D., Malone, R. E.,, and Symington, L.
S. (1991) in The Molecular and Cellular Biology of the Yeast
Saccharomyces. Genome Dynamics, Protein Synthesis, and Energetics
(Brach, J., Jones, E., and Pringle, J., eds.) Cold Spring Harbor Press,
Cold Spring Harbor, pp. 407-521.
28. McEachern, M. J., and Blackburn, E. H. (1996)
Genes Dev., 10, 1822-1834.
29. Murnane, J. P., and Yu, L.-C. (1993) Mol.
Cell. Biol., 13, 977-983.
30. Flint, J., Craddock, C. F., Villegas, A.,
Bentley, D. P., Williams, H. J., Galanello, R., Cao, A., Wood, W. G.,
Ayyub, H., and Higgs, D. R. (1994) Am. J. Hum. Genet.,
55, 505-512.
31. Kramer, K. M., and Haber, J. E. (1993) Genes
Dev., 7, 2345-2356.
32. Biessmann, H., Mason, J. M., Ferry, K., d'Hulst,
M., Valgeirsdottir, K., Traverse, K. L., and Pardue, M.-L. (1990)
Cell, 61, 663-673.
33. Biessmann, H., Valgeirsdottir, K., Lofsky, A.,
Chin, C., Ginther, B., Levis, R. W., and Pardue, M.-L. (1992) Mol.
Cell. Biol., 12, 3910-3918.
34. Mason, J. M., and Biessmann, H. (1995) Trends
Genet., 11, 58-62.
35. Pardue, M. L., Danilevskaya, O. N., Lowenhaupt,
K., Slot, F., and Traverse, K. L. (1996) Trends Genet.,
12, 48-52.
36. Sheen, F., and Levis, R. W. (1994) Proc.
Natl. Acad. Sci. USA, 91, 12510-12514.
37. Levis, R. W., Ganesan, R., Houtchens, K., Tolar,
L. A., and Sheen, F.-M. (1993) Cell, 75, 1083-1093.
38. Moore, J. K., and Haber, J. E. (1996)
Nature, 383, 644-646.
39. Teng, S.-C., Kim, B., and Gabriel, A. (1996)
Nature, 383, 641-644.
40. Lingner, J., Hughes, T. R., Shevchenko, A.,
Mann, M., Lundblad, V., and Cech, T. R. (1997) Science,
276, 561-567.
41. Marcand, S., Gilson, E., and Shore, D. (1997)
Science, 275, 986-990.
42. Van Steensel, B., and de Lange, T. (1997)
Nature, 385, 740-743.
43. Krauskopf, A., and Blackburn, E. H. (1996)
Nature, 383, 354-357.
44. Kyrion, G., Boakye, K. A., and Lustig, A. J.
(1992) Mol. Cell. Biol., 12, 5159-5173.
45. Conrad, M. N., Wright, J. H., Wolf, A. J., and
Zakian, V. A. (1990) Cell, 63, 739-750.
46. Wotton, D., and Shore, D. (1997) Genes
Dev., 11, 748-760.
47. Bryan, T. M., Marusic, L., Bacchetti, S., Namba,
M., and Reddel, R. R. (1997) Hum. Mol. Genet., 6,
921-926.
48. Pereira-Smith, O. M., and Smith, J. R. (1988)
Proc. Natl. Acad. Sci. USA, 85, 6042-6046.
49. Whitaker, N. J., Kidston, E. L., and Reddel, R.
R. (1992) J. Virol., 66, 1202-1206.
50. Duncan, E. L., Whitaker, N. J., Moy, E. L., and
Reddel, R. R. (1993) Exp. Cell Res., 205, 337-344.
51. Klein, H. L. (1995) BioEssays,
17, 147-159.
52. Stürzbecher, H.-W., Donzelmann, B.,
Henning, W., Knippschild, U., and Buchhop, S. (1996) EMBO J.,
15, 1992-2002.
53. Shen, Z., Pardington-Purtymun, P. E., Comeaux,
J. C., Moyzis, R. K., and Chen, D. J. (1996) Genomics,
37, 183-186.
54. Greenwell, P. W., Kronmal, S. L., Porter, S. E.,
Gassenhuber, J., Obermaier, B., and Petes, T. D. (1995) Cell,
82, 823-829.
55. Lustig, A. J., and Petes, T. D. (1986) Proc.
Natl. Acad. Sci. USA, 83, 1398-1402.
56. Kojis, T. L., Gatti, R. A., and Sparkes, R. S.
(1991) Cancer Genet. Cytogenet., 56, 143-156.
57. Metcalfe, J. A., Parkhill, J., Campbell, L.,
Stacey, M., Biggs, P., Byrd, P. J., and Taylor, A. M. R. (1996)
Nature Genet., 13, 350-353.
58. Meyn, M. S. (1993) Science, 260,
1327-1330.
59. Sprung, C. N., Bryan, T. M., Reddel, R. R., and
Murnane, J. P. (1997) Mutat. Res., in press.
60. Szostak, J. W., Orr-Weaver, T. L., Rothstein, R.
J., and Stahl, F. W. (1983) Cell, 33, 25-35.
61. Orr-Weaver, T. L., and Szostak, J. W. (1983)
Proc. Natl. Acad. Sci. USA, 80, 4417-4421.