Reconstruction of Mitochondrial Cytochrome P450-Dependent Steroid Hydroxylases in Artificial Phospholipid Membranes: Studies of Cytochrome P450scc Topology by Limited Proteolysis
A. V. Krivosheev1 and S. A. Usanov1,2
1Institute of Bioorganic Chemistry, Academy of Sciences of Belarus, ul. Zhodinskaya 5, Minsk, 220141 Belarus; fax: (375-172) 63-7274; E-mail: usanov@ns.iboch.ac.by2To whom correspondence should be addressed.
Submitted December 9, 1996; revision submitted June 5, 1997.
Highly purified cytochrome P450scc from bovine adrenal cortex mitochondria was inserted in artificial phospholipid membranes prepared from phosphatidylcholine to study the main principles of its membrane organization in the model system. Topology of the cytochrome P450scc polypeptide chain in proteoliposomes was studied by limited proteolysis with trypsin or chymotrypsin followed by immunochemical identification of the products of proteolysis products of the membrane-bound heme protein. It is shown that limited proteolysis of cytochrome P450scc in proteoliposomes results in a significant decrease of Vmax for the reaction of cholesterol hydroxylation to pregnenolone in the reconstituted system in the presence of exogenously added adrenodoxin-reductase and adrenodoxin. However, after proteolytic modification of cytochrome P450scc with trypsin and chymotrypsin the affinity of the heme protein to adrenodoxin is increased. Different models of membrane organization as well as functional specificity of cytochrome P450scc in artificial membranes are discussed.
KEY WORDS: cytochrome P450scc, artificial membranes, limited proteolysis.
Abbreviations: SDS-PAGE) electrophoresis in polyacrylamide gel in the presence of SDS; AD) adrenodoxin; ADR) adrenodoxin-reductase.
Cytochrome P450-dependent monooxygenase systems of adrenocortical
mitochondria catalyze the key reactions of steroid hormone biosynthesis
[1, 2]. Cholesterol side chain
cleavage cytochrome P450scc is an integral membrane protein which is
localized in the inner mitochondrial membrane and catalyzes the main
reaction of cholesterol transformation to form pregnenolone, which is
thought to be the first and rate limiting step in the process of
steroid hormone biosynthesis. Because of its principal functional role,
cytochrome P450scc is one of the most intensively studied
representative of the whole cytochrome P450 superfamily. However,
despite numerous studies, the molecular organization of cytochrome
P450scc in the inner membrane of adrenocortical mitochondria is studied
considerably less than that of cytochrome P450 from liver endoplasmic
reticulum membranes [3, 4].
Several research groups have carried out series of works devoted to
preparation and characterization of artificial membrane structures
containing cytochrome P450scc as a model system to study the topology
of cytochrome P450scc in the inner membrane of adrenocortical
mitochondria [5-7]. Moreover,
limited proteolysis in combination with immunochemical methods of
identification of proteolysis products have been used many times to
study the membrane organization of cytochrome P450scc in the natural
and in artificial membranes [8-11]. By using this approach it was shown that
cytochrome P450scc, in contrast to cytochromes P450 from endoplasmic
reticulum membranes (which are bound with the membrane only through a
small fragment of the N-terminal sequence), is tightly connected with
the inner mitochondrial membrane of adrenal cortex and has an membrane
matrix-side exposed fragment of polypeptide chain near
Arg250-Asn257. The proteolytic cleavage of this
fragment of the polypeptide chain of membrane-bound cytochrome P450scc
results in formation of fragments F1 and F2, which represent the N- and
C-terminal amino acid sequences of the heme protein, which are,
nevertheless, remaining tightly bound with the membrane. Moreover, for
membrane-bound cytochrome P450scc, in contrast to soluble heme
proteins, there is an alternative pathway for proteolysis that results
in formation of fragment F0; this indicates that this fragment of the
polypeptide chain close to Arg92 is exposed on the membrane
surface.
Proteolytic modification with trypsin followed by immunochemical identification of the reaction products was successfully used by some research groups to study the topology in mitochondrial membrane of cytochrome 45011beta [12] and microsomal steroidogenic cytochrome P450c21 from bovine adrenal cortex [13-15] in natural membranes and phospholipid vesicles as well as membrane topology of some isozymes of cytochrome P450 from liver endoplasmic reticulum membranes [16-19]. Nevertheless, some aspects of the problem of membrane organization of mitochondrial and microsomal cytochromes P450, such as (1) the tightness of the interaction of the heme protein with the membrane and the number of the protein--membrane contacts, (2) the location of different cytochrome P450 fragments relative to the membrane, (3) the presence or absence of transmembrane fragments in the cytochrome P450 molecule able to span the phospholipid membrane and the number of such fragments, if there are any, (4) the location of the substrate-binding center relative to the membrane, (5) the location of the sites responsible for interaction with electron transfer partners, (6) the similarity or distinctions of the principles of molecular organization of mitochondrial and microsomal cytochrome P450 isozymes, remain to be discussed.
The present work opens the cycle of the structure-functional studies of membrane-bound heme proteins and is devoted to further studies of membrane topology of cytochrome P450scc polypeptide chain and its structure-functional peculiarities in artificial membranes. The main objective of the present work was the preparation and use of the simplest model membrane from egg phosphatidylcholine to study the membrane topology of cytochrome P450scc by analyzing the peculiarities of the limited proteolysis of cytochrome P450scc in the artificial membrane and in solution as well as to investigate the effect of trypsin and chymotrypsin on the catalytic activity of the membrane-bound heme protein and on the character of its interaction with adrenodoxin in proteoliposomes. The results obtained provide additional information to extend our conceptions on the structure-functional peculiarities of the membrane-bound cytochrome P450scc.
MATERIALS AND METHODS
Chemicals. In the present work the following reagents were used: Tris, sodium cholate, sodium dodecyl sulfate, Serva Blue R-250, chymotrypsin, soybean trypsin inhibitor, Tween-20, cholesterol, deoxycorticosterone, and glucose-6-phosphate (Serva, Germany); glucose-6-phosphate-dehydrogenase (Fermentas, Lithuania); trypsin (Worthington, USA); nitrocellulose (Schleicher and Schuell, Germany); cholesterol-oxidase (Wako Pure Chemicals Ind., Japan); Triton X-100 (Loba, Austria); NADP+ (Reanal, Hungary); 4-chloro-1-naphthol (Sigma, USA); matrices for gel filtration (Pharmacia, Sweden). The other chemicals were of the highest purity available.
Protein Purification. Cytochrome P450scc, adrenodoxin (AD)3, and adrenodoxin-reductase (ADR) were purified from bovine adrenal cortex mitochondria using affinity chromatography on biospecific ligands as previously described [20]. The purified proteins according to SDS-PAGE were homogeneous and did not contain contaminations. Cytochrome P450scc concentration was determined spectrophotometrically using molar absorption coefficient epsilon450 = 91 mM-1·cm-1 for the carbon monoxide difference spectrum of the sodium dithionite reduced heme protein [21]. The concentrations of AD and ADR were determined spectrophotometrically using molar absorption coefficients epsilon414 = 11.0 mM-1·cm-1 [22] and epsilon450 = 11.3 mM-1·cm-1 [23], respectively.
Preparation of Artificial Membranes. Proteoliposomes were prepared by the gel filtration method [5] using 25 mM sodium phosphate buffer, pH 7.4, containing 0.1 M sodium chloride, 0.1 mM EDTA, and 0.1 mM dithiothreitol. Phosphatidylcholine from egg yolk in chloroform was evaporated under an argon flow and solubilized in buffer solution containing sodium cholate (the maximal final detergent concentration was 1%). Cytochrome P450scc (cytochrome P450/phospholipid ratio, 1:2.8 w/w) was added to solubilized phosphatidylcholine and the reaction mixture was incubated 15 min at 4°C. After the incubation the reaction mixture was subjected to chromatography on a Sephadex G-50 (fine) column (1 × 14 cm) equilibrated with the working buffer.
The integrity of the proteoliposome membranes was monitored by reduction of incorporated potassium ferricyanide. In this case, 0.7 M potassium ferricyanide was added to the mixture of phosphatidylcholine and cytochrome P450scc before application to the Sephadex G-25 column. The reduction of potassium ferricyanide was carried out in the presence of 0.1 mM NADPH and ADR at molar ratio cytochrome P450/ADR 1:0.5. The integrity of the proteoliposome membranes was calculated as a ratio of amount of reduced potassium ferricyanide before and after solubilization of proteoliposomes with 0.3% Triton X-100 and was found to be 90%.
Enzymatic Reduction of Cytochrome P450scc. Enzymatic reduction of cytochrome P450scc was carried out in the reconstituted system containing ADR and AD at the following final concentrations of the proteins: 1) in solution (0.67 µM cytochrome P450scc, 3.34 nM ADR, 0.33-1.67 µM AD); 2) in proteoliposomes (0.67 µM cytochrome P450scc, 3.34 nM ADR, 0.067-1.0 µM AD). After saturation of the sample cell with CO, cytochrome P450scc was reduced by NADPH-generating system containing 5 µM glucose-6-phosphate, 0.5 IU glucose-6-phosphate-dehydrogenase, and 100 µM of NADP+. The increase of optical density at 450 nm was monitored using the option of time scanning.
Limited Proteolysis of Cytochrome P450scc. Limited proteolysis of cytochrome P450scc with trypsin and chymotrypsin was carried out at 25°C using enzyme/substrate weight ratio 1:25. The trypsinolysis was stopped by adding soybean trypsin inhibitor taken at 3-fold weight excess to trypsin. In the experiments on determination of enzymatic activity of cytochrome P450scc in the proteoliposomes the inhibitor was not used. In this case, as well as during the treatment of the proteoliposomes with chymotrypsin, control experiments were done to determine the effect of proteoliposome treatment with chymotrypsin on the functional properties of ADR and AD.
Cytochrome P450scc fragments which formed during its limited proteolysis were separated by SDS-PAGE in 12% polyacrylamide gel [24] and analyzed by immunoblotting [25] using monospecific antibodies against cytochrome P450scc or fragments of its polypeptide chain [9]. Nitrocellulose replicas were incubated in the presence of conjugate of sheep IgG labeled with peroxidase and diluted 1:1000 against rabbit IgG. The unbound conjugate was washed out and the filters were stained in 10 mM sodium phosphate buffer, pH 7.2, containing 0.15 M NaCl, 3.37 mM 4-chloro-1-naphthol, and 10 mM hydrogen peroxide.
Determination of Enzymatic Activity of Cytochrome P450scc. Enzymatic activity of cytochrome P450scc was determined as previously described [26, 27], with some modifications. The protein concentrations in the reconstituted system were: cytochrome P450scc, 1.0 µM; ADR, 0.5 µM; AD, 0.5-10.0 µM. The reaction mixture contained 0.05% Tween-20 and 25 µM cholesterol in the final volume 0.5 ml. The reaction was started by adding the NADPH-generating system, carried out at 25°C, and stopped by putting the tubes into a boiling water bath for 30 sec. The reaction mixture afterwards was incubated with cholesterol-oxidase to convert cholesterol and pregnenolone into cholestenone and pregnenolone as previously described [26, 27]. The reaction was started by the addition to cooled incubation mixture of 50 µl cholesterol-oxidase (0.2 IU) in 20 mM potassium phosphate buffer, pH 7.4, containing 0.3% sodium cholate. After 10 min incubation at 37°C, steroids were extracted with 2.0 ml of either petroleum ether or ethyl acetate. The extracts were evaporated under a nitrogen flow and then dissolved in the mobile phase for HPLC just before analysis. The steroids were separated by normal phase HPLC using an Altex (USA) instrument in isocratic mode (column 4.6 × 250 mm; Lichrosorb, particle diameter, 7 µm; mobile phase, mixture of n-hexane and isopropyl alcohol, 82:18 v/v; rate, 0.9 ml/min). Deoxycorticosterone was used as an internal standard. Chromatograms were analyzed at 254 nm using a Chromatopac C-R1B Shimadzu digital integrator (Shimadzu, Japan).
Spectrophotometric measurements were carried out using Specord M-40 (Germany) and Shimadzu UV-3000 (Japan) spectrophotometers. Electrophoregram scanning was done using a scanning device of our own production suitable for a Specord-UV-VIS (Germany).
RESULTS
For quantitative and qualitative estimation of the degree of inclusion of cytochrome P450scc into proteoliposomes prepared from egg yolk phosphatidylcholine as well as to characterize the prepared proteoliposomes, gel filtration on Sepharose 4B was used. As follows from Fig. 1, the peak on the elution profile corresponding to proteoliposomes has a little less elution volume than "free" cytochrome P450scc; this indicates effective incorporation of the heme protein into the proteoliposomes. To determine more precisely the amount of cytochrome P450scc incorporated into the proteoliposomes and to concentrate proteoliposome preparations, proteoliposomes were centrifuged in Amicon microconcentrators. Since microconcentrator membranes used in our experiments have an exclusion limit of about 100 kD, cytochrome P450scc monomers not included into proteoliposomes were easily separated from proteoliposomes. Moreover, it may be concluded that the main part of the cytochrome P450scc in proteoliposomes is really embedded into the membrane and does not form microaggregates captured inside the proteoliposomal membrane since more than 90% of cytochrome P450scc incorporated into proteoliposomes, as will be shown subsequently, is accessible to trypsin, which is not able to pass through the phospholipid membrane of proteoliposomes. The amount of cytochrome P450scc incorporated into proteoliposomes was 85-90% of the total amount of the initial heme protein.
As follows from the data presented in Table 1, functionally active cytochrome P450scc (based on the carbon monoxide difference spectra of chemically reduced cytochrome P450scc) is 60-65% of the total amount of cytochrome P450scc incorporated into the proteoliposomes 10 min after their preparation. Then, slow inactivation of cytochrome P450scc expressed by its conversion into an inactive form, cytochrome P420, takes place. The content of the later form is about half of the total amount of cytochrome P450scc after 60 min of incubation either at 4 or 25°C. Upon incubation of proteoliposomes during 48 h only 25% of the cytochrome P450scc is retained in its active form.Fig. 1. Gel filtration of proteoliposomes containing cytochrome P450scc (1) and free cytochrome P450scc (2) on Sepharose 4B. Column dimensions, 1.2 × 60.0 cm; elution rate, 3 ml/h. Insert: carbon monoxide difference spectra of reduced cytochrome P450scc in proteoliposomes before (1) and after (2) passing through membranes of an Amicon microconcentrator.
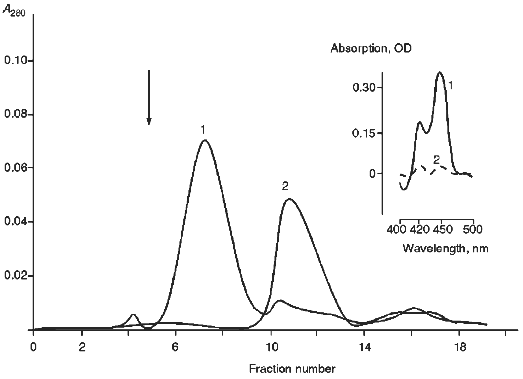
TABLE 1. Dynamics of Inactivation of
Cytochrome P450scc Incorporated in Proteoliposomes and Change of the
Proportion of the Heme Protein Able to be Reduced in the Presence of
ADR, AD, and an NADPH-Generating System (the initial liposomes were
incubated at 4°C)

In the experiments on the enzymatic reduction of cytochrome P450scc in the proteoliposomes in the presence of exogenously added AD and ADR, with subsequent chemical reduction of the heme protein with sodium dithionite as a control, it has been shown that in this case only 40-55% of the active heme protein is enzymatically reduced (Table 1). As mentioned earlier, incubation of proteoliposomes at 4°C during 48 h results in conversion to an inactive form (cytochrome P420) of up to 75% of the cytochrome P450scc incorporated into the membrane. In this case, more than 80% of the functionally active cytochrome P450scc is enzymatically reduced.
To characterize protein--protein interactions with participation of either free or proteoliposome-membrane-incorporated cytochrome P450scc, the kinetic parameters of enzymatic reduction of the heme protein on the dependence of the molar excess of exogenously added AD were measured. During electron transfer AD forms highly specific complexes with ADR and cytochrome P450scc that finally results in electron transfer from NADPH to cytochrome P450scc and reduction of the heme iron of the heme protein. The rate constants for cytochrome P450scc reduction as well as the Michaelis constants (Km) for AD for soluble and membrane-bound cytochrome P450scc in the reaction of its enzymatic reduction in the reconstituted system were calculated (Table 2). The main difference between the two studied systems consists of the different physical state of the terminal oxidase--cytochrome P450scc. Thus, since in the case of membrane-bound cytochrome P450scc we used soluble, exogenously added ADR and AD, the main difference between the two systems consists of the different nature of the interaction of AD with the soluble and membrane-bound cytochrome P450scc. The data presented in Table 2 indicate that in the case of proteoliposomes containing cytochrome P450scc, the process of heme protein reduction is less effective and is characterized by decreased affinity of AD to immobilized cytochrome P450scc; this may be due to the limited mobility and accessibility of membrane incorporated heme protein for AD.
TABLE 2. Kinetic Parameters of Enzymatic
Reduction of Soluble and Membrane-Bound Cytochrome P450scc

To study the molecular organization of cytochrome P450scc in proteoliposomes, we used limited proteolytic modification of the heme protein with trypsin and chymotrypsin. This approach was successfully used previously to study the domain organization of soluble cytochrome P450scc [28] and localization of the heme-containing fragment of the polypeptide chain [29]. In the experiments on treatment of cytochrome P450scc-containing proteoliposomes with trypsin, several polypeptide fragments were obtained which proved to be very similar according to their molecular weight to the fragments which are usually formed during limited proteolysis of soluble cytochrome P450scc.
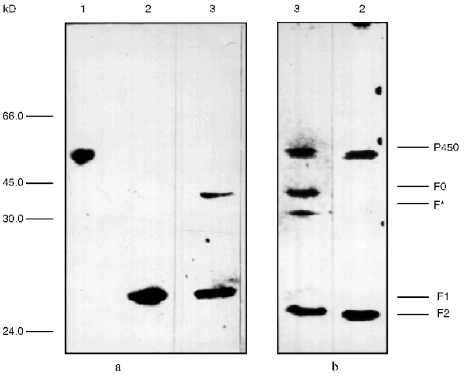
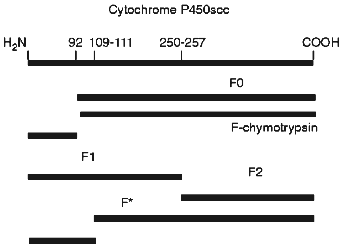
To identify the antigenic specificity and assign the fragments to corresponding structural elements of the cytochrome P450scc molecule, they were subjected to immunoblotting in the presence of specific antibodies against fragments which are formed during limited proteolytic modification of the soluble cytochrome P450scc. The fragments investigated based on their immunochemical properties proved to be very similar to the fragments F1 (Ile1-Arg256), F2 (Asn257-Ala481), and F0 (Arg92-Ala481) (Fig. 2). The fragments F1 (29.8 kD) and F2 (26.6 kD) represent the N- and C-terminal sequences of cytochrome P450scc, respectively, but fragment F0 (45.3 kD) appears to be truncated from the N-terminal sequence (deltaIle1-Arg92) of the cytochrome P450scc polypeptide chain. Fragment F2 in turn can be further degraded with trypsin with formation of fragment F3 (16.8 kD), whereas F3 was never observed in the experiments with proteoliposomes. The formation of the above-mentioned fragments from cytochrome P450scc incorporated into proteoliposomes prepared from phosphatidylcholine shows the surface localization of accessible-to-proteases fragments of the cytochrome P450scc molecule even after its incorporation into the membrane.
Somewhat surprising was the detection of a minor fragment with molecular weight of about 30 kD, further indicated as fragment F* (Fig. 2). The nature and origin of this fragment of cytochrome P450scc polypeptide chain still remains unclear, although the presence of this fragment as a minor fraction shows that this is not the main pathway of proteolytic fragmentation of cytochrome P450scc. However, since fragment F* is detected only by antibodies against fragment F2, this indicates that this fragment has antigenic structure very close to fragment F2, and that this fragment is formed as a result of proteolytic cleavage of a Lys109-Asp111 bond.Fig. 2. Western-blotting with antibodies against fragments F1 (a) and F2 (b) of free cytochrome P450scc (1) and peptide fragments formed after treatment with trypsin of free (2) and membrane-bound (3) cytochrome P450scc. The time of trypsinolysis was 30 min.
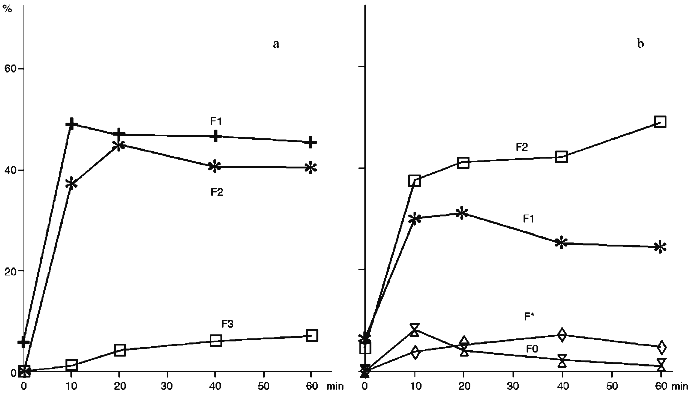
The treatment of proteoliposomes containing cytochrome P450scc with trypsin results in an evident decrease of the content of the N-terminal part (fragment F1) during incubation, while in control series of experiments using soluble cytochrome P450scc, fragment F1 exhibits similar to fragment F2 stability to further degradation by trypsin (Fig. 3). As follows from Fig. 3, trypsinolysis of cytochrome P450scc in proteoliposomes does not result in further proteolytic cleavage of the C-terminal fragment near Arg399, indicating that this fragment of cytochrome P450scc in proteoliposomes is not accessible to trypsin. In contrast, during trypsinolysis of the soluble heme protein the formation of the corresponding fragment F3 is usually observed (Fig. 3a).
Modification of cytochrome P450scc-containing proteoliposomes with chymotrypsin results in formation of only one main fragment with molecular weight about 45 kD. Similar results were obtained for both membrane-bound and soluble cytochrome P450scc (Fig. 4). This polypeptide fragment is "recognized" by antibodies against fragments F1 and F2, thus proving the presence in its structure of the elements of the polypeptide chain which are involved in formation of antigenic determinants both in fragment F1 and fragment F2. Since antibodies against fragments F1 and F2 do not possess cross-reactivity, this means that this fragment contains structural elements arising both from fragment F1 and fragment F2.Fig. 3. Dynamics of peptide fragment formation during trypsin treatment of free (a) and membrane-bound (b) cytochrome P450scc. The total absorption of all stained bands at 650 nm is taken as 100%.
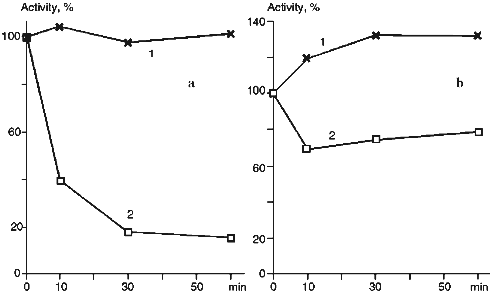
The treatment of proteoliposomes with trypsin for 10 min results in loss of about 40% of the initial enzymatic activity of cytochrome P450scc in the reaction of cholesterol transformation into pregnenolone (Fig. 5a), while the activity of the soluble cytochrome P450scc after such treatment remains at the control level. This is followed by decrease of the Km value for adrenodoxin in the reaction of cholesterol transformation into pregnenolone in proteoliposomes from 0.428 µM (in control experiments) to 0.130 µM (after limited trypsinolysis) (Table 3). After 60 min of limited trypsinolysis, the activity of cytochrome P450scc exhibits only 20% of the control values. Figure 5b shows the effect of the treatment of cytochrome P450scc-containing proteoliposomes with chymotrypsin on its ability to perform cholesterol side chain cleavage. The inhibition of hydroxylation activity in this case consists of not more than 25% during the first 10 min and then the level of activity is 80% of the control value. The value of Km for AD in the case of proteoliposomes differs insignificantly. The limited proteolytic modification of soluble cytochrome P450scc results in 25-30% increase of its catalytic activity.Fig. 4. Western-blotting analysis of proteoliposomes containing cytochrome P450scc (1) and the products of proteolytic modification with chymotrypsin using antibodies against cytochrome P450scc (1, 2) and its fragments F1 (3) and F2 (4). The time of treatment was 30 min.
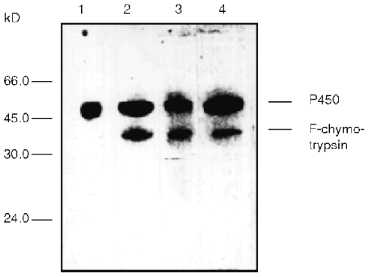
TABLE 3. Effect of Proteolytic Modification with Trypsin and Chymotrypsin on Cholesterol Side Chain Cleavage Activity of Soluble and Membrane-Bound Cytochrome P450sccaFig. 5. Effect of proteolytic modification with trypsin (a) and chymotrypsin (b) on catalytic activity of soluble (a) and membrane-bound (b) cytochrome P450scc. The initial activity of the heme protein before modification is taken as 100%.

DISCUSSION
The results obtained in the present work on spontaneous inactivation of cytochrome P450scc in proteoliposomes prepared from phosphatidylcholine during storage are in accordance with results described in [7], where it is stated that fast and low phases of inactivation of cytochrome P450scc occur when the protein is incorporated into phosphatidylcholine liposomes prepared by gel filtration method. We also found an increase of the amount of cytochrome P450scc retaining the ability to be reduced by electrons from NADPH in the presence of exogenously added ADR and AD during the inactivation process. Thus, after 30 min of incubation about 57.4% of the native heme protein (cytochrome P450scc) is reduced in the reconstituted system, while after 48 h of incubation the enzymatic reduction is to more than 80% of the heme protein which is in the cytochrome P450 form. These results indicate that our proteoliposome preparations contain at least two different modes of incorporation of cytochrome P450scc in the phospholipid membrane; they are characterized by different stability and spatial orientation of the heme protein relative to the phospholipid membrane. Moreover, only part of the cytochrome P450scc in proteoliposomes is able to interact with partner proteins and mainly with AD. This suggestion is supported by the formation of minor amounts of fragment F* during proteolytic modification of membrane-bound cytochrome P450scc that is not characteristic for trypsinolysis of the soluble heme protein.
The authors of the paper [5] did not observe the enzymatic reduction of cytochrome P450scc in proteoliposomes prepared from synthetic dioleoylphosphatidylcholine not containing cholesterol, but inclusion of up to 7.3 mole % of cholesterol in the membrane allowed reduction up to 70% of the cytochrome P450scc. It is reasonable to suggest that in our preparations of proteoliposomes in the absence of exogenously added cholesterol, cytochrome P450scc is present in the membrane in more labile and less aggregated state. It is well known, that substrate serves as a powerful stabilizing agent for cytochrome P450scc; it affects the conformation and spin state of the heme iron.
The preparations of proteoliposomes obtained in the present work according to the absolute absorption spectrum of membrane-incorporated cytochrome P450scc contain the heme protein predominantly in the low spin state, i.e., cytochrome P450scc in proteoliposomes prepared from phosphatidylcholine does not contain cholesterol in the active site. It is reasonable to suggest that a cholesterol molecule is displaced from the active site of the cytochrome P450scc during proteoliposome preparation due to better solubility of cholesterol in the lipid phase. This, however, does not prevent enzymatic reduction of the membrane-bound cytochrome P450scc in the presence of exogenously added ADR and AD. However, it is not possible to exclude the situation when a cholesterol molecule is retained in the active site of cytochrome P450scc but just changes its spatial orientation in such a way that only the hydrophobic side chain of the molecule is displaced from the active site, resulting in the changes in the absorption spectrum of cytochrome P450scc in proteoliposomes [31]. In other words, not the entire cholesterol molecule but only its hydrophobic side chain is removed from the active site resulting in transition of the heme iron of cytochrome P450scc to the low spin state. The latter suggestion is supported by the absence of changes in the optical density at cytochrome P450scc on titration with cholesterol in proteoliposomes (data not shown).
As follows from the data presented in Table 2, the rate of enzymatic reduction (Vmax) of cytochrome P450scc in proteoliposomes in the reconstituted system in the presence of exogenously added ADR and AD is 2.33 times less than that of for the soluble heme protein. It has been shown that an apparent Km which characterizes the interaction of AD with membrane-incorporated cytochrome P450scc increased from 0.087 to 0.145 µM. Therefore, being incorporated in proteoliposomes cytochrome P450scc possesses lower affinity to exogenous AD than the soluble heme protein. This state of cytochrome P450scc may be explained by immobilization of the heme protein in phospholipid structures that decrease the probability and efficiency of interaction with ferredoxin molecules. On the other side, cytochrome P450scc incorporated into membrane may change its conformation resulting in weakening of the interaction of cytochrome P450scc with exogenously added AD. The later results in increase of Km value and simultaneously decreases the rate of cytochrome P450scc reduction from 0.917 to 0.393 nmole/min.
The picture of the limited proteolysis of cytochrome P450scc in proteoliposomes suggests that the N-terminal sequence (fragment F1) of the heme protein molecule in this case is localized on the outside surface of the vesicle membrane, i.e., it is exposed and thus may be partially subjected to proteolysis at an additional sites that results in degradation of some of the fragment F1 not according to the classical scheme, with formation of low molecular weight peptides which are usually not detected by SDS-PAGE. The C-terminal sequence of cytochrome P450scc (fragment F2) appears to be localized in the liposomal membrane or span the phospholipid membrane and be practically inaccessible to proteolytic treatment.
Figure 6 shows a scheme of the most probable sites in the cytochrome P450scc polypeptide chain in proteoliposomes which are accessible to proteolytic enzymes used in the present work. The formation of fragment F0 during limited proteolysis of membrane-bound cytochrome P450scc which is characteristic for limited trypsinolysis of soluble inactivated form of cytochrome P450scc--cytochrome P420--to some extent may be explained by the presence of this form in preparations of proteoliposomes containing cytochrome P450scc. However, the fact that the content of fragment F0 of the total amount of peptide formed does not exceed 15%, but the amount of cytochrome P420 reaches 35-40%, suggests that formation of the fragment F0 may be due to accessibility to protease action of a site close to Arg92 in the native cytochrome P450scc in proteoliposomes. Moreover, this fact also indicates different conformation states of the cytochrome P450scc molecule in the soluble state and in proteoliposomes, since fragment F0 is formed only under treatment with trypsin of soluble cytochrome P420. Furthermore, as has been shown by us previously [9], during limited proteolysis of cytochrome P450scc in submitochondrial particles with trypsin, in contrast to limited proteolysis of soluble cytochrome P450scc, together with usually formed fragments F1 and F2, some amount of fragment F0 is also formed; this cannot be explained by the presence of the inactivated form of cytochrome P450scc in the inner mitochondrial membrane of mitochondria, but reflects the mode of heme protein organization in membrane. This fact indirectly supports the fact of cytochrome P450scc insertion into phospholipid membrane and indicates a similarity of the modes of molecular organization of the heme protein in artificial and natural membranes. The presence of the minor fragment F* can also be explained by a specific organization of a small part of the heme protein in which an atypical polypeptide fragment of cytochrome P450scc located close to residues Lys109-Asp111 (which is normally not accessible to proteolytic modification in soluble cytochrome P450scc and the heme protein in the natural membranes) becomes accessible to trypsin.
The results of limited proteolysis studies of cytochrome P450scc incorporated in proteoliposomes indicates non-equivalent distribution of the N- and C-terminal sequences of cytochrome P450scc in the liposomal membrane and more expressed interaction with the membrane of fragment F2. The latter is in accordance with earlier published data [32] about the presence of prolonged alpha-helical fragments of the polypeptide chain mainly in the fragment F2 of cytochrome P450scc; these might be responsible for the tight interaction of the heme protein with the membrane. This suggestion, however, does not agree with the results presented in paper [33] from which it follows that after limited proteolysis of submitochondrial particles, cytochrome P450scc fragment with molecular weight 26 kD, i.e., fragment F2 according to our terminology, moves to the solution after treatment of the membrane with 50 mM Na2CO3; this indicates that this fragment is not tightly bound with the membrane. This conclusion is also not supported by our results obtained earlier on immunochemical investigation of the topology of cytochrome P450scc in adrenocortical mitochondria using isotopically labeled antibodies against different cytochrome P450scc fragments; this clearly indicates transmembrane orientation of fragment F2 in the membrane [10] as well as the results of the present work indicating that fragment F2 is practically inaccessible to proteolytic modification with trypsin and is presumably localized in the proteoliposomal membrane or spans the phospholipid bilayer.Fig. 6. Scheme of the proposed sites of trypsin attack in cytochrome P450scc molecule incorporated in liposomes during limited proteolysis.
The formation of the main fragment with molecular weight of about 45 kD after treatment of proteoliposomes with chymotrypsin should occur by the cleavage of the full-length heme protein approximately 90-100 amino acid residues from the N-terminal amino acid. Taking into account the data on the primary structure of cytochrome P450scc [34, 35] and based on the fact that the main product of proteolytic modification contains antigenic determinants specific for antibodies against both fragments F1 and F2, it is reasonable to suggest that the polypeptide fragment close to Arg92-Tyr93-Tyr94 is accessible to chymotrypsin and hence exposed on the surface of the liposomal membrane. The cholesterol side chain activity of proteoliposomes insignificantly decreased after treatment with chymotrypsin. We suggest that the cleavage of cytochrome P450scc incorporated in proteoliposomes in the indicated site does not result in serious conformational changes that might affect the substrate-binding properties of the heme protein or the character of its interaction with partner proteins during monooxygenation.
Of significant interest is the inhibition of the catalytic properties of liposomal cytochrome P450scc by trypsin. The decrease of enzymatic activity with concomitant decrease of Km value may be explained by changes of conformational mobility of cytochrome P450scc in the process of trypsinolysis that might result in increased affinity of cytochrome P450scc to AD, on one hand, and weakening of the enzyme--substrate interactions on the other.
The treatment of cytochrome P450scc incorporated into proteoliposomes with proteases does not result in dramatic changes in the independent heme protein inactivation time course described above. The changes of catalytic activity of cytochrome P450scc appear to be the result of more rigid fixation of the heme protein in proteoliposomes, in which the possibility of spontaneous redistribution and aggregation of proteolytic products into functionally active complexes, as occurs under limited proteolysis of soluble cytochrome P450scc, is excluded [20]. Thus, the possible changes of functional behavior of the heme protein during limited proteolysis may be a result of conformational changes in the modified protein.
The results obtained in the present work support the suggestion made earlier of complicated transmembrane organization of cytochrome P450scc and a significant role of the phospholipid microenvironment for the functional state of the heme protein in the membrane. However, the results of the present work indicate a different character of the effects of the proteolytic modification with different proteases on the functional activity of soluble and membrane-bound cytochrome P450scc as well as on a significant role of the fragments Arg250-Arg256 and Lys109-Asp111 in retention of conformational and functional stability of cytochrome P450scc in the artificial membranes studied. The limited proteolysis in combination with immunochemical identification of the products of proteolytic modification of cytochrome P450scc used in the present work as the main methodological approach proved to be very promising for further detailed study of the membrane topology of cytochrome P450scc as well as analysis of protein--protein interactions between the components of the cholesterol side chain cleavage system of adrenal cortex mitochondria and is now intensively used to study membrane topology of cytochrome P450scc in proteoliposomes prepared from different types of phospholipids.
The authors thank Dr. M. A. Kissel for supplying us with highly purified egg yolk phosphatidylcholine and Dr. A. A. Chernogolov for supplying antibodies against cytochrome P450scc and its fragments and for critical reading of our manuscript.
LITERATURE CITED
1.Shkumatov, V. M., Usanov, S. A., Chashchin, V. L.,
and Akhrem, A. A. (1985) Pharmazie, 40, 757-766.
2.Usanov, S. A., Chashchin, V. L., and Akhrem, A. A.
(1990) in Molecular Mechanisms of Adrenal Steroidogenesis and
Aspects of Regulation and Application (Ruckpaul, K., and Rein, H.,
eds.) Frontiers in Biotransformation, Vol. 3, Akademie-Verlag,
Berlin, pp. 1-57.
3.Nelson, D. R., and Strobel, H. W. (1988) J.
Biol. Chem., 263, 6038-6050.
4.Black, S. D. (1993) FASEB J., 6,
680-685.
5.Seybert, D. W., Lancaster, J. R., Lambeth, J. D.,
and Kamin, H. (1979) J. Biol. Chem., 254,
12088-12098.
6.Hall, P. F., Watanuke, M., and Hamkalo, B. A.
(1979) J. Biol. Chem., 254, 547-552.
7.Yamakura, Y., Kido, T., and Kimura, T. (1981)
Biochim. Biophys. Acta, 649, 343-354.
8.Usanov, S. À., Chernogolov, À. À.,
Petrashin, À. I., Akhrem, À. À., and Chashchin, V. L.
(1987) Biol. Membr. (Moscow), 4, 1102-1116.
9.Usanov, S. A., Chernogolov, A. A., and Chashchin,
V. L. (1989) FEBS Lett., 255, 125-128.
10.Usanov, S. A., Chernogolov, A. A., and Chashchin,
V. L. (1990) FEBS Lett., 275, 33-35.
11.Roby, K. F., Larsen, D. L., Deb, S., and Soares,
M. J. (1991) Mol. Cell. Endocrinol., 79, 13-20.
12.Lombardo, A., Laine, M., Defaye, G., Monnier, N.,
Guidicelli, C., and Chambaz, E. M. (1986) Biochim. Biophys.
Acta, 863, 71-81.
13.Ohta, Y., Kawato, S., Tagashira, H., Takemori,
S., and Kominami, S. (1992) Biochemistry, 31,
12680-12687.
14.Kominami, S., Tagashira, H., Ohta, Y., Yamada,
M., Kawato, S., and Takemori, S. (1993) Biochemistry,
32, 12935-12940.
15.Guzov, V. M., Zel'ko, I. N., Bylinskaya, S. A.,
and Usanov, S. A. (1994) Biochemistry (Moscow), 59,
1149-1163 (Russ.).
16.De Lemos-Chiarandini, C., Frey, A. B., Sabatini,
D. D., and Kreibich, G. (1987) J. Cell Biol., 104,
209-219.
17.Tsokos, D. C., Omata, Y., Robinson, R. C.,
Krutzsch, H. C., Gelboin, H. V., and Friedman, F. K. (1992)
Biochemistry, 31, 7155-7159.
18.Laurenzana, E. M., Sorrels, S. L., and Owens, S.
M. (1995) Drug Metab. Dispos., 23, 271-278.
19.Black, S. D., and Martin, S. T. (1994)
Biochemistry, 33, 6945-6951.
20.Usanov, S. À., Pikuleva, I. À.,
Chashchin, V. L., and Akhrem, A. A. (1984) Bioorg. Khim.,
10, 32-45.
21.Omura, T., and Sato, R. (1964) J. Biol.
Chem., 239, 2370-2378.
22.Huang, J. J., and Kimura, T. (1973)
Biochemistry, 12, 406-409.
23.Hiwatashi, A., Ichikawa, Y., Maruya, N., Yamano,
T., and Aki, K. (1976) Biochemistry, 15, 3082-3090.
24.Laemmli, U. K. (1970) Nature, 227,
680-685.
25.Towbin, H., Staehelin, T., and Gordon, J. (1979)
Proc. Natl. Acad. Sci. USA, 76, 3116-3120.
26.Sugano, S., Morishima, N., Ikeda, H., and Horie,
S. (1989) Analyt. Biochem., 182, 327-333.
27.Takeshima, M., and Hara, T. (1991) Biochem.
Biophys. Res. Commun., 179, 161-169.
28.Chashchin, V. L., Shkumatov, V. M., Usanov, S.
A., Vasilevsky, V. I., Turko, I. V., and Akhrem, A. A. (1985)
Biomed. Biochim. Acta, 44, 665-677.
29.Chashchin, V. L., Turko, I. V., Akhrem, A. A.,
and Usanov, S. A. (1985) Biochim. Biophys. Acta, 825,
313-324.
30.Demel, R. A., Jord, W., Lambrechts, H., Van
Damme, H., Hovius, R., and Den Krujiff, B. (1989) J. Biol.
Chem., 264, 3988-3997.
31.Hasumi, H., Yamakura, F., Nakamura, S., Suzuki,
K., and Kimura, T. (1984) Biochim. Biophys. Acta, 787,
152-157.
32.Akhrem, A. A., Adamovich, T. B., Lapko, V. N.,
Sherman, S. A., Usanov, S. A., Shkumatov, V. M., and Chashchin, V. L.
(1985) in Cytochrome P450. Biochemistry, Biophysics, and Induction
(Vereczkey, L., and Magyar, K., eds.) Akademiai Kiado, Budapest,
pp. 113-119.
33.Ou, W., Ito, A., Morohashi, K., Fujii-Kuriyama,
Y., and Omura, T. (1986) J. Biochem., 100,
1287-1296.
34.Chashchin, V. L., Lapko, V. N., Adamovich,
O. B., Lapko, A. G., Kuprina, N. S., Kirillova, N. I.,
Berikbaeva, O. I., Akhrem, A. A., and Zolotarev, A. I.
(1985) Bioorg. Khim., 11, 1048-1067.
35.Morohashi, K., Yoshioka, H., Gotoh, O., Okada,
K., Yamamoto, K., Miyata, T., Sogawa, K., Fujii-Kuriuama, Y., and
Omura, T. (1984) Proc. Natl. Acad. Sci. USA, 81,
4647-4651.